Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Patricia Moreira and Version 2 by Catherine Yang.
Alzheimer’s disease (AD) is the most common neurodegenerative disorder affecting elderly people worldwide. Currently, there are no effective treatments for AD able to prevent disease progression, highlighting the urgency of finding new therapeutic strategies to stop or delay this pathology. Several plants exhibit potential as source of safe and multi-target new therapeutic molecules for AD treatment. Meanwhile, Eucalyptus globulus extracts revealed important pharmacological activities, namely antioxidant and anti-inflammatory properties, which can contribute to the reported neuroprotective effects.
- Alzheimer’s disease
- E. globulus
1. Aβ Formation and Tau Hyperphosphorylation
According to the “Amyloid Cascade Hypothesis”, accumulation and oligomerization of Aβ peptide in the brain plays a major role in AD pathophysiology [1][131]. Aβ is a short fragment formed by the amyloidogenic proteolytic cleavage of the amyloid precursor protein (APP) [2][132] (Figure 12), which exhibits toxic effects on neuronal and glia cells in both oligomeric and fibrillar forms. Therefore, several approaches have been designed to decrease Aβ peptide formation from APP, and the most studied targets are β-secretase (BACE) and the γ-secretase complex. APP cleavage is performed by these two enzymes at variable sites to form numerous fragments of Aβ [3][4][133,134]. There are two isoforms of BACE [2][132]: BACE-1 [5][135] and BACE-2 [6][136]. The inhibition of BACE-1 is the most attractive therapeutic approach in AD because Aβ production from APP cleavage in the brain mainly results from the action of this β-secretase isoform. The membrane fragment formed upon BACE1 action is then cleaved by γ-secretase, generating Aβ fragments, namely Aβ1-40 and Aβ1-42 [7][137]. The inhibition of γ-secretase is also a valuable strategy but is less attractive than β-secretase due to fact that it is a multiprotein complex. Unfortunately, serious side effects were revealed in the clinical trials performed with secretase inhibitors [8][9][138,139] since the inhibition of these two enzymes can interfere with the processing of other substrates [10][11][140,141].
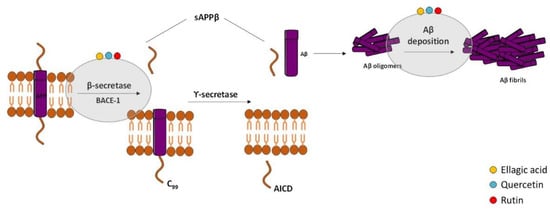
Figure 12. Effect of compounds obtained from E. globulus leaves in the amyloidogenic pathway and in the formation of amyloid-β (Aβ) in AD. The amyloidogenic pathway is initiated with the enzymatic breakdown of amyloid precursor protein (APP) by β-secretase enzyme followed by catalytic cleavage of APP by γ-secretase to originate non-soluble protein or Aβ. Aβ oligomerization and accumulation leads to synaptic dysfunction and neurodegeneration.
Hyperphosphorylated tau-enriched NFTs are another neuropathological hallmark of AD. Under physiologic conditions, tau is the principal microtubule (MT)-associated protein that cooperates with tubulin to regulate MTs stability, which is crucial to axonal transport and thus to neuronal functioning [12][143]. In AD, hyperphosphorylated tau loses the capacity to bind MTs and forms NFTs that contribute to the neurodegenerative process [13][14][144,145] (Figure 23). Overproduction of inflammatory mediators has been shown to activate kinases such as cyclin-dependent kinase-5 (CDK-5) and glycogen synthase kinase-3β (GSK-3β), which consequently lead to tau phosphorylation [15][16][146,147]. In AD, GSK-3β plays a crucial role in tau hyperphosphorylation [17][148], but it was also demonstrated to contribute to Aβ aggregation and deposition into senile plaques [18][149]. With this in mind, GSK-3β inhibitors could represent a promising treatment strategy for AD.
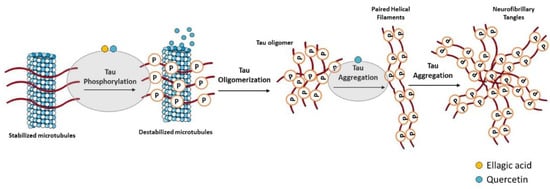
Figure 23. Effect of compounds obtained from E. globulus leaves on tau aggregation and formation of neurofibrillary tangles (NFTs) in AD. Irregular phosphorylation of tau proteins destabilizes microtubules, leading to the formation of insoluble tau oligomers, which then accumulate to generate protomers. Then, two twisted protomers originate paired helical filaments, which after aggregation lead to the formation of NFTs. These intracellular structures are involved in synaptic and neuronal dysfunction, thus contributing to cognitive decline in AD.
In the last years, some studies revealed that phenolic compounds can interfere with both amyloid and tau pathologies, supporting their beneficial role in AD. However, there is no information in the literature about the effect of EO from E. globulus leaves and its major compound 1,8-cineole on AD. Ellagic acid was found as a potential BACE-1 inhibitor as well as a protective strategy against Aβ deposition and tau hyperphosphorylation. In a screening for anti-dementia agents from natural products, Kwak and collaborators (2005) reported that ellagic acid was a moderate BACE-1 specific inhibitor [19][150] and in vitro studies showed that ellagic acid promoted a significant loss of oligomers levels and was able to prevent Aβ-induced toxicity [20][21][151,152]. Accordingly, ellagic acid treatment in a sporadic AD rat model induced by streptozotocin (STZ) administration markedly decreased brain Aβ levels, suggesting its potential to delay amyloidogenesis [22][153]. Finally, it was reported that ellagic acid decreased APP and BACE-1 expression levels as well as Aβ deposition in the hippocampus of APP/PS1 transgenic mice, a model of familial AD [23][154]. This study also described the inhibition of tau hyperphosphorylation by ellagic acid mediated by the activation of the protein kinase B (Akt)/GSK-3β signaling pathway.
2. Oxidative Stress
The presence of oxidative stress markers in the AD brain has been pointed out as another relevant AD hallmark. Oxidative stress is caused by an imbalance between the production of reactive oxygen species (ROS) and/or reactive nitrogen species (RNS) and the removal capacity of the antioxidant system, promoting damage to lipids, proteins, ribonucleic acids (RNA), and deoxyribonucleic acids (DNA) [24][25][179,180]. Despite the mechanisms by which the redox balance is altered in AD and the sources of ROS/RNS remain unknown, numerous studies suggest that Aβ is a potent trigger of oxidative stress that is, at least in part, mediated by the disruption of mitochondrial function and subsequent generation of oxidant species [26][181] (Figure 34). Therefore, development of novel antioxidant strategies is required to prevent AD progression.
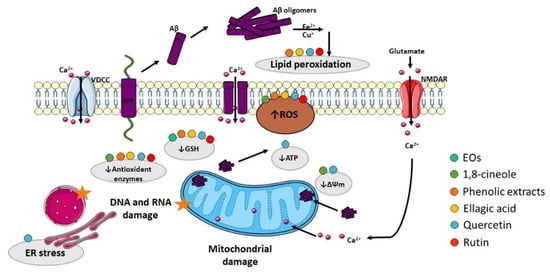
Figure 34. Effect of compounds obtained from E. globulus leaves in oxidative stress and mitochondrial damage in AD. Aβ oligomers can insert the plasma membrane originating pores by which Ca2+ pass into the cytoplasm. Aβ can also interact with metal ions (Fe2+ and Cu+) to generate reactive oxygen species (ROS), which cause membrane lipid peroxidation. As consequence, the membrane turns depolarized, and voltage-dependent Ca2+ channels (VDCC) and glutamate receptor-associated channels (in particular NMDAR, N-methyl-D-aspartate receptor) open increasing cytoplasmic Ca2+ content. Additionally, Aβ overproduction can cause mitochondrial damage, which culminates in ROS accumulation and ATP depletion that can impair axonal transport consequently originating abnormal mitochondrial dynamics and promoting neurotransmission deficits. ATP depletion can also lead to ionic alterations in the cytosol due to dysfunction of ATP-dependent ion channels. Moreover, ROS accumulation affects the mitochondrial permeability transition pore (MPTP), which further potentiates mitochondrial damage due to Ca2+ overload and inhibition of the electron transport chain. ROS increase also promotes damage to proteins, namely DNA and RNA.
Many studies reported the antioxidant properties of EOs from E. globulus, which contribute to its neuroprotective effects [27][28][17,116]. For example, an in vitro study performed by Mizuno (2015) found that hydrogen peroxide (H2O2)-induced neuronal death was attenuated by the EO of E. globulus [29][182]. Moreover, Yadav (2019) showed that E. globulus oil alleviated depressive and cognitive symptoms of ketamine-induced psychosis in rats mediated by its antioxidant effect in the cerebral cortex and hippocampus, where the levels of reduced glutathione (GSH) were restored [30][77]. In both studies, the reported protective effect of EO from E. globulus might be due to the presence of 1,8-cineole, which was shown to be the major component. In fact, Ryu (2014) showed that 1,8-cineole may attenuate oxidative stress in cortical neuronal/glial cells through its antioxidant capacity as ROS scavenger and activator of superoxide dismutase (SOD) [31][183]. Additionally, an in vitro study using a neuronal cell model, performed by Khan and colleagues in 2014, demonstrated that Aβ-induced neuronal toxicity was prevented by 1,8-cineole pretreatment. The loss of mitochondrial membrane potential as well as ROS accumulation were attenuated by 1,8-cineole, supporting its anti-oxidative properties [32][184]. On the other hand, as observed above in the previous section, α-pinene is also present in the EO of E. globulus leaves and in vivo studies revealed its antioxidant effect. Lee (2017) demonstrated that α-pinene increased protein levels of antioxidant enzymes, namely the heme oxygenase-1 (HO-1) and manganese superoxide dismutase (MnSOD) in the hippocampus of the scopolamine-induced AD mice model via activation of the nuclear factor erythroid 2-related factor 2 (Nrf2) [33][185], which is a transcription factor that stimulates an antioxidant defense response. Nrf2 levels decrease with age, and reduced Nrf2 levels were reported in AD animal models and postmortem human brain tissue from patients [34][186]. Interestingly, recent studies revealed that Nrf2 activators may delay the progression and ameliorate the symptoms of the disease, suggesting that Nrf2 inducers might be relevant therapeutic molecules for AD [35][187].
González-Burgos (2018) investigated the antioxidant activity of different extracts (acetone, ethanol, and methanol) from E. globulus leaves and concluded that the extracts rich in phenolic compounds were effective to prevent H2O2-induced oxidative stress and preserve cell viability, increasing the activity of antioxidant enzymes and GSH levels as well as decreasing lipid peroxidation and ROS production in SH-SY5Y cells [36][13]. As mentioned before, the ellagic acid is one of the most predominant compounds found in phenolic extracts from E. globulus leaves. In fact, several studies reported the antioxidant properties of ellagic acid with significant impact on the progression of AD pathology, particularly through the activation of several antioxidant enzymes, reducing lipid peroxidation and free radical scavenging activity. Kabiraj and collaborators (2014) showed that ellagic acid is able to scavenge peroxynitrite, protecting PC12 cells against rotenone-induced cell death and also to reduce ROS and RNS production in these neuronal-like cells. Moreover, these authors demonstrated that ellagic acid suppressed apoptosis caused by rotenone by reducing poly (ADP-ribose) polymerase-1 (PARP) cleavage, which is a hallmark of apoptotic cell death [37][188].
Quercetin and its glycoside rutin are two abundant compounds found in phenolic extracts of eucalyptus leaves, and several in vitro and in vivo studies have investigated their neuroprotective potential in AD. Both compounds were reported to attenuate oxidative stress in different AD models, mainly by decreasing ROS production and lipid peroxidation and increasing GSH content and the activity of several antioxidant enzymes. In APPswe cells, which are a cellular model of AD consisting of cells transfected with Swedish mutated human APP, Jimenez-Aliaga and collaborators (2011) demonstrated that quercetin and rutin decrease ROS generation and lipid peroxidation and increase intracellular GSH content, improving the redox status of APPswe cells treated with H2O2 [38][155]. In addition, rutin and quercetin were found to have free radical scavenging activity and to ameliorate Aβ-induced neuronal death in mouse primary cortical neuronal cultures [39][193]. Moreover, rutin attenuated mitochondrial damage and reduced the levels of ROS and oxidized glutathione (GSSG) as well as the formation of MDA and stimulated the activity of the antioxidant enzymes CAT, SOD, GSH, and glutathione peroxidase (GPx) in microglia cells exposed to Aβ [40][156]. Rutin was also demonstrated to inhibit amylin-induced neurotoxicity in SH-SY5Y cells, reducing the formation of ROS, GSSG, and MDA; attenuating mitochondrial damage and increasing the GSH/GSSG ratio; and enhancing the antioxidant activity of SOD, CAT, and GPx [41][194]. Additionally, quercetin was shown to preserve cell viability in PC12 cells treated with H2O2 [42][195]. An in vitro study with primary hippocampal cultures described that low doses of quercetin significantly attenuated Aβ-induced cytotoxicity, lipid peroxidation, protein oxidation, and apoptosis; however, higher dosages were reported to potentiate neuronal dysfunction [43][196]. Later studies demonstrated that quercetin protected rat primary hippocampal neurons against H2O2- or Aβ-induced neurotoxicity, attenuating ROS accumulation and depolarization of the mitochondrial membrane [44][197]. The role of quercetin in OA-induced oxidative stress in HT22 hippocampal cells was investigated, and it was found that pre-treatment with quercetin activates SOD, avoids GSH depletion, and decreases ROS production and MDA levels. The alterations in membrane potential caused by OA were reversed by quercetin, further supporting its neuroprotective action [45][164]. Quercetin was also reported to raise intracellular GSH content and prevent oxidative/nitrosative damage to DNA, lipids, and proteins in SH-SY5Y cells exposed to a neurotoxin [46][198]. On the other hand, rutin pretreatment was shown to decrease TBARS and PARP activity and increase GSH content and the activity of GPx, glutathione reductase, and CAT enzymes in the hippocampus of rats treated with STZ [47][199]. The effect of rutin was investigated in APPswe/PS1dE9 transgenic mice, and it was demonstrated that it decreased GSSG and MDA levels and increased SOD activity and GSH/GSSG ratio [48][178]. Moreover, lipid peroxidation was decreased in the brain, liver, and kidneys by treatment with rutin in an AD mouse model induced by Aβ injection [49][200]. Oxidative damage was attenuated by rutin treatment in rats with chronic cerebral hypoperfusion, namely GPx activity were increased, and the levels of MDA and protein carbonyls were decreased in rutin-treated animals [50][201]. Furthermore, it was recently demonstrated that pretreatment with rutin reduced CAT, GSH, and SOD protein levels in rats injected with doxorubicin [51][202]. Furthermore, an in vivo study performed by Tota and collaborators showed that quercetin restored cerebral blood flow and adenosine triphosphate (ATP) content after STZ administration in mice and reduced oxidative and nitrosative stress as demonstrated by a reduction in MDA and by an increase in GSH content [52][203]. It was also reported that quercetin treatment reduced MDA levels in the brain of STZ-induced diabetic rats [53][204]. In addition, quercetin decreased MDA generation in brain homogenates of mice treated with trimethyltin and showed strong antioxidant capacity determined through free radical scavenging activity assays [54][205]. Furthermore, lipid peroxidation was shown to be significantly inhibited by quercetin in the brain of Aβ-injected mice [55][206]. Indeed, increased SOD, CAT, and GSH and decreased MDA levels were observed in the brain of Aβ-injected rats treated with quercetin, concomitantly with activation of the antioxidant Nrf2/HO-1 pathway [56][177]. Quercetin ameliorated mitochondrial dysfunction, as evidenced by restoration of mitochondrial membrane potential and ROS and ATP levels in mitochondria isolated from the hippocampus of APP/PS1 transgenic mice. Furthermore, the activity of AMPK, which is a master regulator of cellular energy and metabolism, was significantly increased by quercetin [57][170]. Recent studies demonstrated that quercetin prevented the mitochondrial apoptotic pathway and neuronal degeneration by a mechanism involving regulation of BAX/Bcl2 and reduction of caspase-3 activity, cytochrome c release, and PARP cleavage in the brain of mice treated with lipopolysaccharide (LPS) [58][207]. Finally, reduction of MDA levels in animals injected with Aβ by rutin and quercetin were associated with upregulation of cAMP-response element binding protein (CREB) and brain-derived neurotrophic factor (BDNF) [59][60][208,209], which is an important regulator of neuronal growth and synaptic plasticity. CREB is one of the essential regulators of BDNF since its phosphorylated form binds to a specific sequence in the BDNF promoter and controls its transcription [61][210].
Several evidences support a crosstalk between oxidative stress and endoplasmic reticulum (ER) stress. In AD, the accumulation of misfolded proteins in susceptible brain regions suggests that the impairment of ER proteostasis machinery is involved in AD pathophysiology [62][211]. Therefore, ER stress can be considered as a therapeutic target for AD treatment. Under conditions of misfolded proteins overload within the ER lumen, ER stress sensors initiate the unfolded protein response (UPR) to reestablish homeostasis. This pathway comprises the activation of three ER trans-membrane proteins, namely inositol-requiring enzyme 1α (IRE1α), PKR-like ER kinase (PERK), and activating transcription factor 6 (ATF6) [61][63][210,212]. IRE1α activation promotes splicing of X-Box-binding protein 1 (XBP1)-mRNA [63][212] and the spliced XBP1 accumulated inside the nucleus upregulates crucial genes to reestablish global proteostasis under ER stress [64][213]. Furthermore, IRE1α can also activate relevant signaling mediators, namely c-Jun N-terminal kinase (JNK), which regulates autophagy and apoptosis [65][214]. ATF6 is an ER-membrane-bound transcription factor that triggers the transcription of ER molecular chaperones [61][210]. PERK also acts as an ER stress sensor, and under stress conditions, the α-subunit of eukaryotic initiation factor 2 (eIF2α) is oligomerized and phosphorylated by PERK [63][212]. This inhibits global protein translation, decreasing the overload of misfolded proteins [66][67][215,216]. Moreover, eIF2α phosphorylation increases translation of the activating transcription factor 4 (ATF4), which encodes genes of autophagy and proteins responsible for cell redox and metabolic regulation [67][216]. In addition, under chronic ER stress, ATF4 upregulates the transcription factor C/EBP homologous protein (CHOP), GADD34, and numerous members of Bcl2 family such as BAX and BAK, two central apoptotic regulators [68][217]. GADD34 can revert the eIF2α phosphorylation in a feed-forward cycle to close PERK signaling [69][218]. There are some evidences that quercetin ameliorates ER stress in AD models. In 2015, Hayakawa and colleagues reported that quercetin can rescue proteostasis, decreasing eIF2α phosphorylation, ATF4 expression, and Aβ secretion through GADD34 upregulation in cells upon autophagy impairment or ER stress conditions, which was confirmed in vivo using an AD mouse model [70][219]. In addition, quercetin repressed ER stress by reducing phosphorylation of eIF2α, PERK, and IRE1α; suppressed oxidative stress by reducing intracellular ROS production; and restored mitochondrial membrane potential in OA-treated SH-SY5Y cells. The same study also reported reduced IRE1α and PERK phosphorylation in mice exposed to high-fat diets [71][167]. A recent study performed by Woo and co-authors in Aβ-injected mice revealed that quercetin attenuates oxidative stress, namely ROS and TBARS generation. Under these conditions, a decrease was observed in the levels of ER stress markers such as phosphorylated eIF2α and PERK, XBP1, and CHOP as well as of pro-apoptotic Bax, phosphorylated JNK, and cleaved caspases-3 and -9 together with upregulation of the anti-apoptotic protein Bcl2 [72][176].
3. Inflammation
Recent evidences suggest that inflammation has a fundamental role in AD pathogenesis; therefore, controlling the interactions between the nervous and the immune system might be crucial to prevent or delay the disease [73][220]. Brain inflammation seems to play a neuroprotective role in acute-phase responses but becomes deleterious during a chronic response to toxic insults [74][221]. Activated microglia release a diversity of proinflammatory and toxic products, including ROS, nitric oxide (NO), and cytokines such as interleukin-1β (IL-1β), interleukin-6 (IL-6), and tumor necrosis factor alpha (TNF-α), which play a significant role in the neuroinflammatory process. Aβ peptide increases the levels of cytokines, including TNF-α and IL-1β, and in turn, elevated levels of IL-1β potentiate Aβ accumulation [73][75][220,222]. Additionally, elevated levels of IL-1β can increase the production of other cytokines, such as IL-6, which activates the CDK-5 kinase that can lead to tau hyperphosphorylation [76][223]. Neuroinflammation has emerged as a third relevant hallmark in AD that can act as a link between amyloid and tau pathologies [77][224] (Figure 45). In fact, immune-related cells and proteins have been reported to be located within close proximity to senile plaques [78][79][225,226], and some evidences indicate that the prolonged use of nonsteroidal anti-inflammatory drugs (NSAIDs) reduce the risk to develop AD and delays the progression of the disease [80][227], possibly due to the inhibition of cyclooxygenases (COX) and activation of peroxisome proliferator-activated receptor γ (PPARγ) [80][227]. COX expression is repressed by NSAIDs, which declines the synthesis of prostaglandins and decreases the secretion of cytokines [81][228]. There are also evidences that NSAIDs decrease the level of Aβ in neuronal cell cultures and transgenic mice modelling AD [80][227]. Nevertheless, additional studies are required to confirm the beneficial effect of NSAIDs in AD.
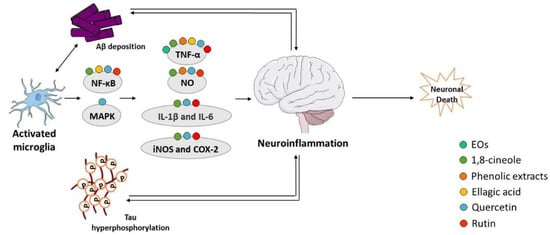
Figure 45. Effect of compounds obtained from E. globulus leaves on neuroinflammation in AD. A vicious circle between Aβ and tau accumulation in the brain, microglia activation, and release of pro-inflammatory cytokines culminates in neuronal death in AD.
E. globulus EO and its major component were recently reported to have anti-inflammatory activity relevant in the AD context. It has been previously reported that the expression of proinflammatory cytokines TNF-α, IL-1β, and IL-6 was lowered by 1,8-cineole in cells exposed to Aβ, and 1,8-cineole also succeeds in reducing NO accumulation and downregulating inducible NO synthase (iNOS), COX-2, and NF-κB [32][184]. More recently, the EO from E. globulus was demonstrated to reduce the serum levels of TNF-α in rats with psychosis, in the absence of any other significant alteration in inflammatory markers [30][77].
There are some evidences that extracts from eucalyptus leaves and ellagic acid reduce inflammation through depletion of TNF-α levels in AD models. Akhtar and collaborators extracted eucalyptus leaves with ethanol and detected anti-inflammatory activity, as shown by inhibition of TNF-α and NO production in macrophages exposed to LPS and interferon-γ (INF-γ) [82][229]. An in vitro study performed in cultured primary murine cortical microglia demonstrated that ellagic acid decreases Aβ-induced TNF-α secretion [83][230]. Another in vivo study showed that the reduction of hippocampal nuclear/cytoplasmatic Nrf2 ratio in Aβ-microinjected rats was reversed by ellagic acid treatment, which also reverted the alterations in NF-κB and TLR4 expression [84][189]. Moreover, ellagic acid was shown to prevent the accumulation of TNF-α detected in the STZ-induced AD rat model [85][86][190,191].
The anti-inflammatory effects of quercetin and rutin in AD models has been reported in several studies, which describe a decrease in NO production and in the expression of proinflammatory cytokines. Regarding in vitro studies, Wang and co-authors observed that rutin reduced NO formation and iNOS activity and also modulated the production of proinflammatory cytokines by decreasing TNF-α and IL-1β generation in microglia cells treated with Aβ [40][156]. Similarly, rutin was showed to reduce the production of NO, iNOS activity, and release of the pro-inflammatory cytokines TNF-α and IL-1β in amylin-treated SH-SY5Y cells, attenuating neurotoxicity [41][194]. Additionally, a study performed in LPS-stimulated microglia cells reported that rutin decreases expression levels of TNF- α, IL-1β, IL-6, and iNOS as well as the secretion of IL-6, TNF-α, and NO and increases the production of interleukin-10 (IL-10), the M2 regulatory cytokine, as well as arginase. Moreover, rutin also restored LPS-induced upregulation of COX-2, interleukin-18 (IL-18), and transforming growth factor-β (TGF-β) [87][231]. Similarly, in vitro studies have also linked quercetin’s neuroprotective effect with its anti-inflammatory activity. For example, quercetin was shown to prevent the release of TNF-α and IL-6 from activated microglia and astrocytes and attenuated the activation of proinflammatory signaling pathways such as MAPK and NF-κB [46][198]. Thioredoxin-interacting protein (TXNIP) is a crucial node in ER stress and NLR family pyrin domain containing 3 (NLRP3) inflammasome, which activates caspase-1, leading to IL-1β secretion to cause inflammation in cells or tissues [88][232]. NLRP3 inflammasome is a protein complex that comprises NLRP3, the adaptor protein apoptosis-associated speck-like protein containing a C-terminal caspase-activation-and-recruitment domain (CARD) (ASC), and the precursor pro-caspase-1. Consistent with this, quercetin suppressed TXNIP expression and NLRP3 inflammasome activation indicated by downregulation of NLRP3, ASC, and procaspase-1 in OA-treated SH-SY5Y cells. Quercetin effectively reduced IL-1β and IL-6 production in neuronal cells and restored NLRP3 activity and reduced IL-1β and TNF-α production in mice exposed to a high-fat diet [71][167]. Quercetin also attenuated neuroinflammation in a mouse model of AD decreasing IL-1β and monocyte chemoattractant protein-1 (MCP-1) levels [89][233]. A study using quercetin-treated 3xTg-AD mice showed a reduction in reactive microglia and astrocytes, glial fibrillary acidic protein (GFAP), iNOS, and COX-2 immunoreactivity as well as IL-1β levels in hippocampal lysates [90][173]. Quercetin also reduced LPS-induced gliosis and the levels of various inflammatory markers, such as TNF-α, COX-2, and iNOS, in the cortex and hippocampus of adult mice [58][207]. Finally, quercetin decreased NO formation in STZ and Aβ- injected mice [52][55][203,206]. In vivo studies with rutin also disclosed its anti-inflammatory activity in AD context. Indeed, rutin ameliorated STZ-induced inflammation in rats by decreasing NO levels and the expression of GFAP, interleukin-8 (IL-8), COX-2, iNOS, and NF-κB [47][199]. Rutin also inhibited glial activation; reduced the levels of pro-inflammatory cytokines TNF-α, IL-1β, and IL-6; and prevented neuronal damage in rats with chronic cerebral hypoperfusion [50][201]. Other study showed that chronic treatment with rutin decreases TNF-α levels in the hippocampus and frontal cortex of rats injected with doxorubicin [51][202]. The oral administration of rutin was also found to downregulate microgliosis and astrocytosis and to reduce IL-1β and IL-6 levels in the brain of the APP/PS1 transgenic AD mice model [48][178]. Additionally, the NO formation was reduced by rutin in Aβ-injected mice [49][200].
4. Cholinesterase Activity
The “Cholinergic Hypothesis” is central to explain AD pathophysiology (Figure 56). This hypothesis considers that cholinergic neurons are affected in AD, leading to a decrease in the synthesis of the neurotransmitter ACh and subsequent release to the synaptic cleft, resulting in cognitive decline and memory loss [91][92][234,235]. Therefore, inhibitors of AChE and butyrylcholinesterase (BChE) enzymes that degrade ACh can represent a therapeutic strategy to increase the levels of ACh in the synaptic cleft and its binding to post-synaptic receptors, thus potentiating cholinergic neurotransmission. In fact, three of the four drugs approved for the relief of AD symptoms are AChE inhibitors, namely donepezil, rivastigmine, and galantamine [93][5]. The active sites of AChE/BChE enzymes bind these cholinesterase inhibitors in a reversible manner and avoid ACh degradation, facilitating cholinergic neurotransmission. Thus, AD symptoms are ameliorated due to the rise of ACh concentration in the synaptic cleft [94][236]. However, the efficacy of cholinesterase inhibitors in AD treatment is limited, and side effects have been reported, such as nausea, abdominal pain, diarrhea, dyspepsia, vomiting, and skin rash [95][237]. Hence, the discovery of new cholinesterase inhibitors from medicinal plant sources concomitantly presenting less adverse effects can be a valuable strategy.
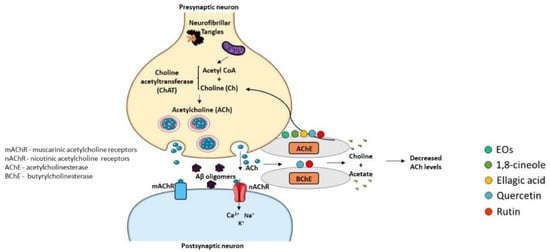
Figure 56. Effect of compounds obtained from E. globulus leaves on cholinesterase activity in AD. Synthesis of acetylcholine (ACh) neurotransmitter from acetyl coenzyme A (Acetyl CoA) and choline (Ch) occurs by the action of the enzyme choline acetyltransferase (ChAT) in the presynaptic terminal. Acetylcholine is released in the synaptic cleft, where it can activate both muscarinic (mAChR) and nicotinic (nAChR) receptors. Acetylcholinesterase (AChE) or butyrylcholinesterase (BChE) break acetylcholine into choline and acetate. ACh levels are low in AD brains and cholinergic neurotransmission in impaired. AChE and BChE inhibitors correct these deficits increasing the amount of ACh that remains in the synaptic cleft and interacts with postsynaptic receptors.
The effective in vitro inhibition of AChE activity by E. globulus EO has been described [96][16]. Moreover, studies in cellular models detected anti-cholinesterase activity of 1,8-cineole and α-pinene [97][98][238,239]. In addition, the AChE inhibitory activity of eucalyptus EO in the hippocampus region of rat’s brain with psychotic symptoms was recently reported [30][77]. Additionally, mRNA levels of enzymes involved in ACh metabolism were evaluated in the cortex of scopolamine-induced amnesic animals, and it was observed that the administration of α-pinene reverted the decrease in the mRNA levels of choline acetyltransferase (ChAT), which is responsible for the formation of ACh [33][185]. However, mRNA levels of AChE were not altered by scopolamine treatment in the presence or absence of α-pinene. These studies revealed the neuroprotective potential of E. globulus EO and its major compounds due to their capacity to inhibit AChE activity.
Ellagic acid was recently described to reduce AChE activity in the brain of animals injected with Aβ [84][189] or with STZ [22][85][153,190], supporting the ability of ellagic acid to reduce cerebral ACh degradation and its neuroprotective role.
Quercetin and rutin also demonstrated to inhibit AChE activity, revealing neuroprotective effects, particularly in AD. In fact, a study of Ademosun and colleagues showed that both compounds significantly decrease AChE and BChE activities in rat brain homogenates, but quercetin showed a higher inhibitory ability than rutin [99][240]. One docking study concluded that rutin exhibited an elevated docking score against AChE in comparison with quercetin, suggesting that rutin is a promising drug candidate for AD [100][241]. Rutin treatment was also found to alleviate ACh depletion and ChAT inhibition as well as the activation of AChE caused by cerebral hypoperfusion in rats [50][201]. On the other hand, in vitro studies demonstrated that quercetin has a strong inhibitory effect against AChE and BChE enzymes [101][102][103][160,242,243], and a relevant role of quercetin as an AChE inhibitor has been described, supporting its therapeutic potential for AD [104][105][106][244,245,246]. Accordingly, several other in vitro studies found similar or higher AchE inhibitory activity of quercetin over conventional AchE inhibitors [107][247]. It was observed that quercetin has significant AChE inhibitory activity almost similar to that of huperzine A [108][248] or donepezil [109][249], which are well-known AChE inhibitors. In addition, these results were confirmed in vivo, and quercetin has been reported to attenuate the AChE activity in the brain of STZ-treated mice [52][53][203,204]. Another study revealed that quercetin suppressed AChE activation in a dose-dependent manner in brain tissues of mice exposed to neurotoxic trimethyltin [54][205]. Finally, Liu and co-authors showed that quercetin was able to restore cortical ACh levels and inhibit AChE activity in Aβ-injected mice [60][209].
5. GABAergic and Glutamatergic Dysfunction
γ-Aminobutyric acid (GABA) is a major inhibitory neurotransmitter in the human brain, which plays a relevant role in cognitive functions [110][250] (Figure 67A). Significant reductions in cerebral GABA levels have been described in AD patients as well as in AD animal models [111][251]. GABAA is one isoform of GABA receptors, and some studies have demonstrated decreased GABAA/benzodiazepine (BZD) receptor density [112][113][252,253] and expression levels [114][254] in the brain of AD patients. Interestingly, the role of selective GABAA agonists to counteract Aβ-induced toxicity was showed [115][255] suggesting that the GABAergic system is involved in the pathophysiology of AD and therefore may be a potential therapeutic target for this neurodegenerative disorder. Recently, it was found that eucalyptus oil increases brain GABA levels [30][77], and α-pinene acts as a partial modulator of GABAA-BZD receptors and binds directly to the BZD binding site of the GABAA receptor [116][256].
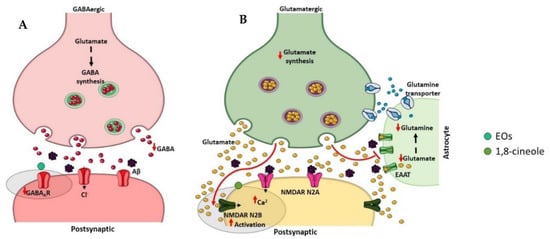
Figure 67. Effect of compounds obtained from E. globulus leaves on inhibitory (A) and excitatory (B) synapses in AD. (A) The inhibitory γ-aminobutyric acid (GABA) synapse. GABA is synthesized from glutamate by the glutamic acid decarboxylase enzymes in the presynaptic terminal of GABAergic neuron. The vesicular GABA transporter packs GABA into vesicles, which, after release in the synaptic cleft, binds GABAA receptors localized on the postsynaptic neuron. The reuptake of GABA into the presynaptic axon stops the GABA action in the synapse. GABA levels are significantly reduced in AD patients as well as the GABAA receptor density. (B) The excitatory glutamate synapse. Glutamine is converted to glutamate via glutaminase in the presynaptic terminal of glutamatergic neuron, and the vesicular glutamate transporter packs glutamate into vesicles. After glutamate release in the synaptic cleft, it acts on glutamate receptors localized on the postsynaptic neuron. The excitatory amino acid transporters (EAATs) present in nearby astrocytes clear the glutamate from the synaptic cleft. Glutamate is converted to glutamine via glutamine synthetase in astrocytes before being transported to presynaptic neurons. In AD, Aβ oligomers affect extrasynaptic N-methyl-D-aspartate (NMDA) receptors enriched in NR2B subunits, leading to an excessive activation and consequently to an excess of Ca2+ accumulation in the post-synaptic cell.
Glutamate is an excitatory neurotransmitter typically present in the hippocampus and cerebral cortex that plays an important role in learning and memory [117][257] (Figure 67B). There are two types of post-synaptic glutamate receptors, ionotropic and metabotropic G protein-coupled receptors, which modulate calcium and sodium influx into neuronal cells [118][258]. However, excessive activation of glutamate receptors, in particular the N-methyl-D-aspartate (NMDA) subtype of ionotropic receptors, provokes excitotoxic neuronal death [119][259]. In AD, an excessive activation of the NMDA receptor has been described and contributes to the neurodegenerative process in consequence of the excessive influx of calcium [119][259]. Numerous evidences suggest that blocking excitotoxicity might be beneficial in AD. Indeed, memantine, which was approved by FDA and EMA for the treatment of AD symptoms, is an uncompetitive NMDA receptor antagonist that blocks excitoxicity with minimal side effects due to the preservation of normal glutamatergic transmission [120][260]. A study using computational models proposed 1,8-cineole as a good candidate for NMDA antagonism comparing its molecular features with the conventional ligand memantine [121][261].
These promising findings suggest that the effect of E. globulus EO on GABAergic and glutamatergic transmission should also be explored as therapeutic strategies for AD. Nevertheless, there is no information in the literature about phenolic compounds and AD-associated perturbation of GABAergic and glutamatergic neurotransmission.
6. Impaired Learning and Memory
Learning is the process of acquiring new information, while memory is the process of storing this information to use it for future purposes (Figure 78). Cognition is defined as the combination of learning and memory and is strictly dependent on the concerted action of several neurotransmitters.
Figure 78. Effect of compounds obtained from E. globulus leaves in the AD-associated memory and learning impairment.
In AD, the stage and severity of the disease are determined by the compromise in cognition [27][17]. The deterioration of cholinergic neurons have been reported to be implicated in cognitive deficits in AD patients [122][262]. Accordingly, anticholinergic agents such as scopolamine have been reported to induce memory deficits [123][263], and on the other hand, an improvement of the cholinergic system can revert alterations in cognition [124][264]. Based on this, and as previously stated, AChE and BChE inhibitors demonstrated to revert cognitive symptoms and have been approved for AD treatment.
EO from E. globulus was recently demonstrated to be able to restore learning and memory function in rats treated with ketamine that induces psychosis [30][77]. In addition, the administration of α-pinene attenuated learning and memory impairments induced in rats treated with scopolamine [33][185].
Several in vivo studies also support the beneficial effects of ellagic acid in cognition of AD animal models. For example, it was demonstrated that ellagic acid efficiently prevents scopolamine- and diazepam-induced cognitive impairments without affecting animals’ locomotion [125][265]. Treatment with ellagic acid also showed to ameliorate memory and spatial learning alterations in the APP/PS1 transgenic AD mice model [23][154]. Moreover, the study of Kiasalari and collaborators described that ellagic acid ameliorates learning and memory performance in Aβ-injected rats [84][189]. Regarding the STZ-induced AD rat model, it was observed that ellagic acid prevents the STZ-induced cognitive deficits in animals without affecting locomotor activity and motor coordination [22][85][86][153,190,191].
Strong evidences support that quercetin and rutin prevent cognitive impairments in several AD animal models. Pretreatment with quercetin and rutin prevented scopolamine-induced memory impairment in zebrafish without locomotor alterations [126][266]. Rutin also ameliorated deficits in learning and memory in STZ rats [47][199] as well as the problems in spatial learning and memory, in working memory, and also in contextual memory in rats with chronic cerebral hypoperfusion [50][201]. In AD transgenic mice, it was demonstrated that rutin decreased spatial memory deficits [48][178] and alleviated cognition and memory impairments in Aβ-injected mice [49][200]. Furthermore, rutin restored short- and long-term episodic memory in scopolamine- and doxorubicin-treated rats without interfering with the locomotor activity of the animals [51][127][202,267]. Quercetin administration in aged and LPS-treated mice also enhanced the memory capacity in the absence of alterations in locomotion [58][128][207,268]. Furthermore, quercetin avoided STZ-induced memory impairment in mice [52][203] and enhanced spatial memory in rats [129][269]. Quercetin also prevented the impairment of memory and the anxiogenic-like behavior induced in STZ-diabetic rats [53][204]. Additionally, quercetin treatment attenuated trimethyltin-induced memory impairment in mice [54][205]. Moreover, in a study with mice exposed to a high-fat diet, quercetin administration enhanced cognition [71][167]. Another study also showed that a quercetin-enriched diet during the early-middle pathology stages ameliorated cognitive dysfunction in APP/PS1 mice [130][175]. In addition, beneficial effects of quercetin in learning, memory deficits, and cognitive function were demonstrated in APP/PS1, APP23, and 3xTg-AD transgenic mice models of AD [57][70][131][132][170,171,172,219]. Furthermore, quercetin administration in Aβ-induced amnesic mice enhanced learning and memory performance [55][60][133][206,209,270]. Finally, two studies with rats injected with Aβ also demonstrated the capacity of quercetin to enhance learning and memory [56][134][177,271]. Importantly, in early-stage AD patients, memory recall assessed using the Revised Hasegawa Dementia Scale was demonstrated to be enhanced by the intake of quercetin [135][272].