Glioblastoma (GBM) is the most lethal primary brain tumor. With limited therapeutic options, novel therapies are desperately needed. Recent studies have shown that GBM acquires large amounts of lipids for rapid growth through activation of sterol regulatory element-binding protein 1 (SREBP-1), a master transcription factor that regulates fatty acid and cholesterol synthesis, and cholesterol uptake. Interestingly, GBM cells divert substantial quantities of lipids into lipid droplets (LDs), a specific storage organelle for neutral lipids, to prevent lipotoxicity by increasing the expression of diacylglycerol acyltransferase 1 (DGAT1) and sterol-O-acyltransferase 1 (SOAT1), which convert excess fatty acids and cholesterol to triacylglycerol and cholesteryl esters, respectively.
- glioblastoma
- fatty acids
- cholesterol
- lipid droplets
1. Introduction
2. Standard GBM Therapy
GBM is a WHO grade IV glioma, with a median survival post-diagnosis of only approximately 15 months despite extensive treatments [23]. GBM represents more than 60% of all brain tumors in adults and occurs in 2–3 cases per 100,000 individuals in Europe and North America [24]. Approximately 90% of GBM cases occur as primary tumors that have wild-type isocitrate dehydrogenase (IDH) and less than three months of clinical symptoms before diagnosis, while less than 5% of all cases are secondary tumors with IDH mutations, progressing from WHO grade II/III gliomas and with better prognosis [12,25,26,27][12][25][26][27]. The standard treatment for GBM is to first perform maximal surgical resection, followed by concomitant radiotherapy and alkylating agent Temozolomide (TMZ) for 6 weeks, and then continuation of TMZ alone every 4 weeks for six cycles of maintenance treatment [28]. Unfortunately, this treatment is only effective for a few months and almost all GBM tumors unavoidably recur after treatment [29]. The 5-year survival for patients with GBM is only around 6.8% in the United States [30]. In the clinic, except for TMZ, only Bevacizumab, a humanized monoclonal antibody against vascular endothelial growth factor (VEGF), has been approved by the FDA for the treatment of recurrent GBM and not for newly diagnosed GBM [31,32][31][32]. It is effective at reducing symptoms and improving the quality of life only for a short time [25]. However, Bevacizumab has no combinatory effect with TMZ and radiotherapy [25]. Moreover, it reduces the uptake of TMZ, fails to increase the overall survival, and has significant side effects such as hypertension, venous thrombosis, and infections [25].3. Lipid Metabolism Regulation in GBM
3.1. Lipogenesis
Glucose is the main source for lipid production through the de novo synthesis pathway [1]. Glucose through glycolysis generates pyruvate that is converted to acetyl-CoA in the mitochondria by the pyruvate dehydrogenase (PDH) [1,39][1][33]. Condensation of acetyl-CoA with oxaloacetate (OAA) forms citrate, which is then released into the cytosol by the SLC25A1 transporter and converted back to acetyl-CoA by ATP-citrate lyase (ACLY). Acetyl-CoA then serves as a precursor for fatty acid and cholesterol synthesis [1,40][1][34] (Figure 1). In addition, acetyl-CoA synthetase 2 (ACSS2) can convert cytosolic acetate to acetyl-CoA for de novo fatty acid and cholesterol synthesis (Figure 1) to promote tumor growth [1]. ACSS2 is upregulated in GBM [41][35] and in hepatocellular carcinoma (HCC), myeloma, prostate, and bladder cancers [42][36]. Furthermore, acetyl-CoA is also involved in epigenetic regulation as it enters into the nucleus to modify histone proteins by direct acetylation [43][37].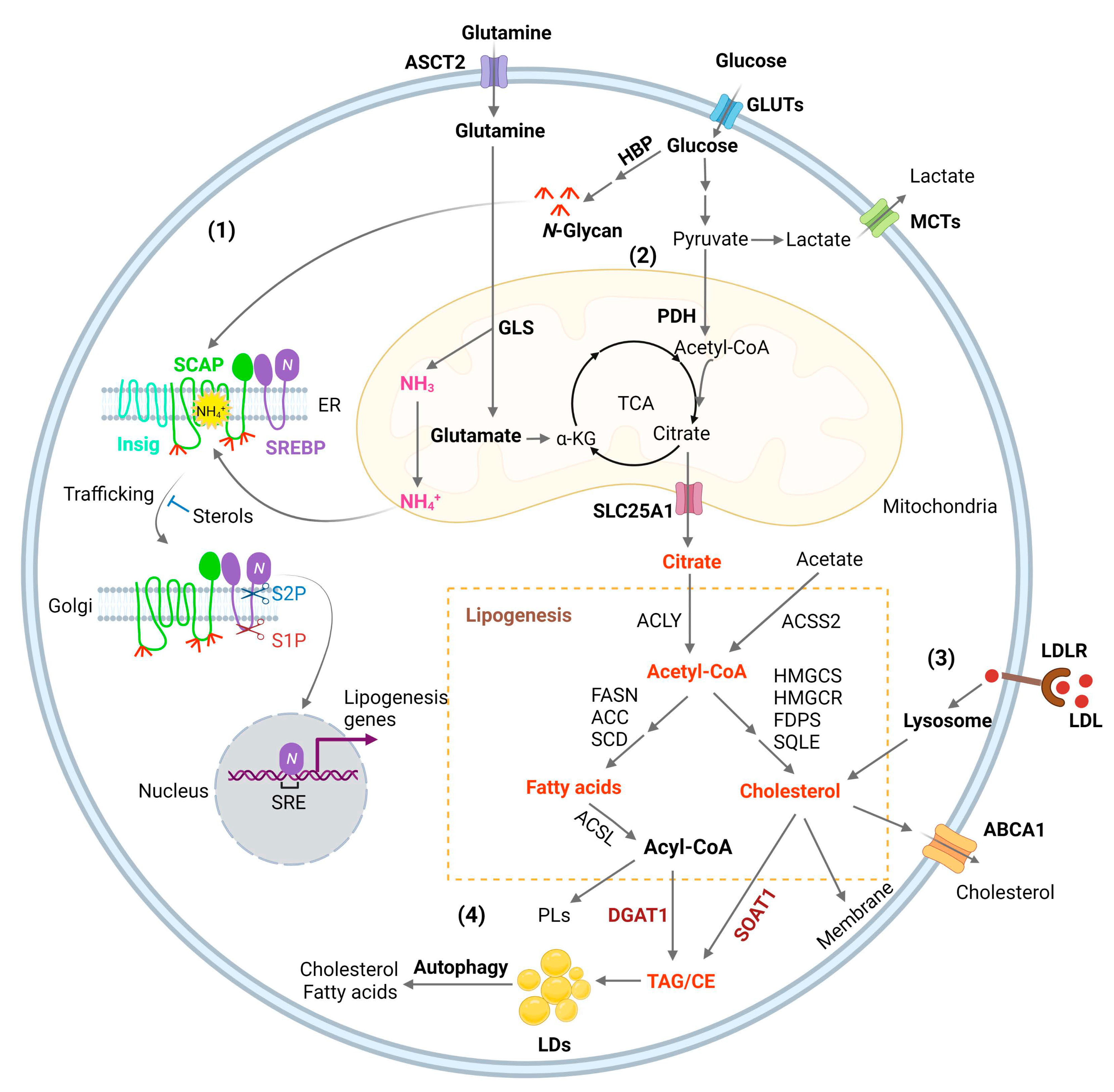
| Target | Inhibitors | Types of Cancer | Preclinical Evidence | Clinical Trials | References |
|---|---|---|---|---|---|
| SREBPs | Fatostatin, Betulin, PF-429242, dipyridamole | GBM, breast, uterine, prostate, liver, lung, kidney, pancreatic and colon |
Xenografts | [1][19][44][45][46][47][48][49][50][51][52][53][54] | |
| ACLY | SB-201076, Hydroxycitric acid, Bempedoic acid, BMS-303141, SB-204990, | Lung, prostate | Xenografts | [1][55][56] | |
| ACCs | ND-646, ND-654, soraphen A, TOFA | Lung, liver, breast | Xenografts, DEN-injured rats |
[17][57][58][59][60][61][62][63][64] | |
| FASN | TVB-2640, C75, cerulenin, orlistat | Lung, colon, breast, GBM, astrocytoma, prostate, uterine | Xenografts | NCT03808558 NCT02223247 NCT02980029 NCT03179904 NCT05118776 NCT03032484 |
[65][66][67][68][69][70][71][72] |
| SCD1 | A939572, MF-438, CAY10566 MK-8245 |
Lung, pancreatic, kidney, GBM, ovarian | Xenograft, Pdx1Cre; LSL-KrasG12D mouse |
NCT00972322 (type 2 diabetes) |
[73][74][75][76][77][78][79][80] |
| ACSS2 | Tetrazoles, Pyridine Derivatives | [81][82] | |||
| SLC25A1 | CTPi2, BTA, CNASB | [83][84] | |||
| CPT1A | Etomoxir, Perhexiline, ST1326 | GBM, breast, leukemia, prostate, colon | Xenografts, Transgenic mice |
[85][86][87][88][89][90][91][92] | |
| DGAT1 | A922500, AZD3988, PF-04620110 |
GBM, liver, uterine, prostate | Xenografts | [17][57][62][63][64][93] | |
| SOAT1 | Avasimibe, ATR-101, K604, | Liver, colon, GBM, kidney | P53-deficient mice, AOM/DSS-treated and ApcMin/+ mice, xenografts | NCT01898715 | [65][66][67][69][70] |
3.2. SREBP Activation, Connecting Glucose and Glutamine to Lipid Synthesis
GBM growth consumes large amounts of glucose and amino acids [56][99], but the mechanism by which tumor cells sense their levels to trigger lipid synthesis has been left unanswered for a long time. OuResearchers' most recent study identified that glucose and glutamine coordinate to activate SREBP-1 to trigger de novo lipid synthesis in GBM and various other cancer cells [57][100]. The SREBP family includes three isoforms, SREBP-1a, -1c, and -2 [58,59,60][101][102][103]. SREBP-1c mainly regulates the expression of genes controlling fatty acid synthesis, while SREBP-2 regulates cholesterol synthesis and uptake, and SREBP-1a, which has the highest transcriptional activity, regulates all three processes [1,61,62,63][1][104][105][106]. SREBPs are synthesized as ~125 kD inactive precursors, which are spatially restrained in the endoplasmic reticulum (ER) membrane and are activated through a tightly controlled ER–Golgi–nucleus translocation process [59,62][102][105]. SREBPs bind to SREBP-cleavage activating protein (SCAP), which further binds to COPII-coated vesicles to move from the ER to the Golgi [62,64][105][107]. In the Golgi, SREBPs are sequentially cleaved by site-1 and -2 proteases to release their N-terminal forms (~65 kD) that then enter the nucleus to activate lipogenic gene expression [65,66,67,68,69][108][109][110][111][112]. Interestingly, SCAP/SREBP trafficking is inhibited by an ER-resident protein, insulin-inducible gene protein (Insig), which includes two isoforms, Insig-1 and -2 [70,71][113][114]. Insig binds to SCAP to retain the SCAP/SREBP complex in the ER (Figure 1) [59,64][102][107]. Cholesterol or 25-hydroxycholesterol (25-HC) can bind to SCAP or Insig to further enhance their association, which represents a negative feedback loop to modulate SREBP activation (Figure 1) [70,72,73][113][115][116]. The key step activating the SCAP/Insig dissociation for subsequent SREBP translocation has just been elucidated in a recent study by Cheng et al. [57][100]. OuResearchers' previous study showed that glucose stimulates SREBP activation and lipogenesis by promoting SCAP N-glycosylation and stability [46,74,75,76][40][117][118][119]. Unexpectedly, ouresearchers' most recent study shows that when glutamine is removed from the medium, glucose alone is unable to activate SREBPs and lipogenesis, even with low cholesterol levels and in the presence of SCAP N-glycosylation [57][100]. WResearchers uncovered an unprecedented role of ammonia, which is released by glutaminolysis and acts as a key activator of the dissociation of N-glycosylated SCAP from Insig by inducing dramatic conformational changes in the SCAP transmembrane domain through interaction via hydrogen bonds with the side chains of three residues, i.e., aspartate D428, serine S326 and S330, eventually leading to SREBP activation and lipid synthesis (Figure 1) [57][100].3.3. Membrane Phospholipid Remodeling
Phospholipids (PLs) constitute the main membrane structure as they form lipid bilayers through their amphiphilic characteristics [77][120]. Elevated fatty acid synthesis promotes PL formation and membrane expansion to facilitate rapid tumor growth [1,78,79][1][121][122]. The composition of the fatty acid chains in PLs is not static, but is dynamically remodeled through the coordinated activities of phospholipase A (PLA) and lysophosphatidylcholine acyltransferases (LPCATs) [80][123]. PLA removes an esterified fatty acid from the sn-2 position of PLs to convert them to lysophosphatidylipids, while LPCATs add back fatty acids to lysophosphatidylipids, thereby remaking PLs [81,82][124][125]. Thus, fatty acid chains in membrane PLs are modified by these two enzymes to adjust to the dynamic alterations of the cellular environment [82][125]. The LPCAT family contains four members (LPCAT1-4) [83,84][126][127]. LPCAT1 mainly regulates the incorporation of saturated fatty acids into lysophosphatidylcholine (LPC) to form PLs, thereby increasing the levels of saturated PLs. Interestingly, LPCAT1 has been shown to be upregulated in various cancers, including GBM [85][128], HCC [86][129], clear cell renal cell cancer [87][130], esophageal squamous cell carcinoma (ESCC) [88][131], gastric [89][132], and breast cancer [90][133]. Its upregulation regulates oncogenic EGFR signaling in GBM by affecting EGFR and its constitutively active mutant EGFRvIII protein stability [85][128]. Targeting LPCAT1 effectively suppresses GBM [85][128] and ESCC growth [88][131]. LPCAT1 elevation across cancer types suggests that the increase in membrane saturated PL levels might prevent lipid peroxidation and ferroptosis in cancer cells, thereby facilitating tumor growth. LPCAT3 regulates the incorporation of polyunsaturated fatty acids, such as arachidonic acid, thereby increasing membrane fluidity and flexibility [84][127]. LPCAT3 has been shown to be a major player to facilitate lipid peroxidation and ferroptosis via increasing membrane polyunsaturated PL level [91,92,93,94][134][135][136][137]. Nevertheless, there has no study clearly demonstrating the expression levels of LPCPAT3 in tumor tissues and its role in tumorigenesis.3.4. Cholesterol Uptake
Cholesterol, which is an indispensable lipid molecule for maintaining cell viability [95][138], is inserted between phospholipid molecules to control membrane integrity and fluidity [96][139], and accounts for 30–40% of membrane lipids in the plasma membrane [97,98,99,100][140][141][142][143]. Cholesterol is esterified with fatty acids to convert to neutral cholesteryl esters (CE), which are carried by low-density lipoprotein (LDL) to circulate in the bloodstream and be absorbed by different tissues [101][144]. LDL is endocytosed into cells as mediated by the LDL receptor (LDLR) and is then delivered to the lysosomes, where LDL is broken down by lysosomal lipases to release free cholesterol, which then egresses from the lysosomes for maintenance and expansion of the plasma membrane but also of the membranes of all cellular organelles such as the mitochondria and endoplasmic reticulum (ER) (Figure 1) [15,102,103,104,105,106][15][145][146][147][148][149].3.5. Lipid storage and Energy Homeostasis
Excess free fatty acids and cholesterol can cause lipotoxicity, leading to cell death when the levels are uncontrolled [107,108][150][151]. In the human body, excess fatty acids and cholesterol are sequestered into the adipocytes of fat tissues within specialized organelles known as lipid droplets (LDs) after being converted to triacylglycerol (TAG) and cholesteryl esters (CE) (Figure 1) [109][152]. Interestingly, in 2016, Geng et al. reported for the first time that LDs are highly prevalent in human GBM tumor tissues, while not observed in normal brain tissues [19,20][19][20]. Using transmission electronic microscopy, they clearly showed that LDs are present in the cytosol of GBM tumor cells.References
- Cheng, C.; Geng, F.; Cheng, X.; Guo, D. Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun. 2018, 38, 27.
- Cockcroft, S. Mammalian lipids: Structure, synthesis and function. Essays Biochem. 2021, 65, 813–845.
- Cisa-Wieczorek, S.; Hernandez-Alvarez, M.I. Deregulation of Lipid Homeostasis: A Fa(c)t in the Development of Metabolic Diseases. Cells 2020, 9, 2605.
- Onal, G.; Kutlu, O.; Gozuacik, D.; Dokmeci Emre, S. Lipid Droplets in Health and Disease. Lipids Health Dis. 2017, 16, 128.
- Estes, R.E.; Lin, B.; Khera, A.; Davis, M.Y. Lipid Metabolism Influence on Neurodegenerative Disease Progression: Is the Vehicle as Important as the Cargo? Front. Mol. Neurosci. 2021, 14, 788695.
- Teixeira, V.; Maciel, P.; Costa, V. Leading the way in the nervous system: Lipid Droplets as new players in health and disease. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2021, 1866, 158820.
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674.
- Snaebjornsson, M.T.; Janaki-Raman, S.; Schulze, A. Greasing the Wheels of the Cancer Machine: The Role of Lipid Metabolism in Cancer. Cell Metab. 2020, 31, 62–76.
- Broadfield, L.A.; Pane, A.A.; Talebi, A.; Swinnen, J.V.; Fendt, S.M. Lipid metabolism in cancer: New perspectives and emerging mechanisms. Dev. Cell 2021, 56, 1363–1393.
- Bian, X.; Liu, R.; Meng, Y.; Xing, D.; Xu, D.; Lu, Z. Lipid metabolism and cancer. J. Exp. Med. 2021, 218, e20201606.
- Cloughesy, T.F.; Cavenee, W.K.; Mischel, P.S. Glioblastoma: From molecular pathology to targeted treatment. Annu. Rev. Pathol. 2014, 9, 1–25.
- Cruz Da Silva, E.; Mercier, M.C.; Etienne-Selloum, N.; Dontenwill, M.; Choulier, L. A Systematic Review of Glioblastoma-Targeted Therapies in Phases II, III, IV Clinical Trials. Cancers 2021, 13, 1795.
- Guo, D.; Hildebrandt, I.J.; Prins, R.M.; Soto, H.; Mazzotta, M.M.; Dang, J.; Czernin, J.; Shyy, J.Y.; Watson, A.D.; Phelps, M.; et al. The AMPK agonist AICAR inhibits the growth of EGFRvIII-expressing glioblastomas by inhibiting lipogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 12932–12937.
- Guo, D.; Prins, R.M.; Dang, J.; Kuga, D.; Iwanami, A.; Soto, H.; Lin, K.Y.; Huang, T.T.; Akhavan, D.; Hock, M.B.; et al. EGFR signaling through an Akt-SREBP-1-dependent, rapamycin-resistant pathway sensitizes glioblastomas to antilipogenic therapy. Sci. Signal. 2009, 2, ra82.
- Guo, D.; Reinitz, F.; Youssef, M.; Hong, C.; Nathanson, D.; Akhavan, D.; Kuga, D.; Amzajerdi, A.N.; Soto, H.; Zhu, S.; et al. An LXR agonist promotes glioblastoma cell death through inhibition of an EGFR/AKT/SREBP-1/LDLR-dependent pathway. Cancer Discov. 2011, 1, 442–456.
- Guo, D.; Cloughesy, T.F.; Radu, C.G.; Mischel, P.S. AMPK: A metabolic checkpoint that regulates the growth of EGFR activated glioblastomas. Cell Cycle 2010, 9, 211–212.
- Cheng, X.; Geng, F.; Pan, M.; Wu, X.; Zhong, Y.; Wang, C.; Tian, Z.; Cheng, C.; Zhang, R.; Puduvalli, V.; et al. Targeting DGAT1 Ameliorates Glioblastoma by Increasing Fat Catabolism and Oxidative Stress. Cell Metab. 2020, 32, 229–242.e8.
- Rudalska, R.; Harbig, J.; Snaebjornsson, M.T.; Klotz, S.; Zwirner, S.; Taranets, L.; Heinzmann, F.; Kronenberger, T.; Forster, M.; Cui, W.; et al. LXRalpha activation and Raf inhibition trigger lethal lipotoxicity in liver cancer. Nat. Cancer 2021, 2, 201–217.
- Geng, F.; Cheng, X.; Wu, X.; Yoo, J.Y.; Cheng, C.; Guo, J.Y.; Mo, X.; Ru, P.; Hurwitz, B.; Kim, S.H.; et al. Inhibition of SOAT1 Suppresses Glioblastoma Growth via Blocking SREBP-1-Mediated Lipogenesis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 5337–5348.
- Geng, F.; Guo, D. Lipid droplets, potential biomarker and metabolic target in glioblastoma. Intern. Med. Rev. 2017, 3.
- Wu, X.; Geng, F.; Cheng, X.; Guo, Q.; Zhong, Y.; Cloughesy, T.F.; Yong, W.H.; Chakravarti, A.; Guo, D. Lipid Droplets Maintain Energy Homeostasis and Glioblastoma Growth via Autophagic Release of Stored Fatty Acids. iScience 2020, 23, 101569.
- Cheng, X.; Geng, F.; Guo, D. DGAT1 protects tumor from lipotoxicity, emerging as a promising metabolic target for cancer therapy. Mol. Cell. Oncol. 2020, 7, 1805257.
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996.
- Rock, K.; McArdle, O.; Forde, P.; Dunne, M.; Fitzpatrick, D.; O’Neill, B.; Faul, C. A clinical review of treatment outcomes in glioblastoma multiforme--the validation in a non-trial population of the results of a randomised Phase III clinical trial: Has a more radical approach improved survival? Br. J. Radiol. 2012, 85, e729–e733.
- Janjua, T.I.; Rewatkar, P.; Ahmed-Cox, A.; Saeed, I.; Mansfeld, F.M.; Kulshreshtha, R.; Kumeria, T.; Ziegler, D.S.; Kavallaris, M.; Mazzieri, R.; et al. Frontiers in the treatment of glioblastoma: Past, present and emerging. Adv. Drug Deliv. Rev. 2021, 171, 108–138.
- Ohgaki, H.; Kleihues, P. The definition of primary and secondary glioblastoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 764–772.
- Li, Z.H.; Guan, Y.L.; Liu, Q.; Wang, Y.; Cui, R.; Wang, Y.J. Astrocytoma progression scoring system based on the WHO 2016 criteria. Sci. Rep. 2019, 9, 96.
- Weller, M.; van den Bent, M.; Tonn, J.C.; Stupp, R.; Preusser, M.; Cohen-Jonathan-Moyal, E.; Henriksson, R.; Le Rhun, E.; Balana, C.; Chinot, O.; et al. European Association for Neuro-Oncology (EANO) guideline on the diagnosis and treatment of adult astrocytic and oligodendroglial gliomas. Lancet Oncol. 2017, 18, e315–e329.
- Ringel, F.; Pape, H.; Sabel, M.; Krex, D.; Bock, H.C.; Misch, M.; Weyerbrock, A.; Westermaier, T.; Senft, C.; Schucht, P.; et al. Clinical benefit from resection of recurrent glioblastomas: Results of a multicenter study including 503 patients with recurrent glioblastomas undergoing surgical resection. Neuro-Oncology 2016, 18, 96–104.
- Ostrom, Q.T.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2014–2018. Neuro-Oncology 2021, 23 (Suppl. 2), iii1–iii105.
- Friedman, H.S.; Prados, M.D.; Wen, P.Y.; Mikkelsen, T.; Schiff, D.; Abrey, L.E.; Yung, W.K.; Paleologos, N.; Nicholas, M.K.; Jensen, R.; et al. Bevacizumab alone and in combination with irinotecan in re-current glioblastoma. J. Clin. Oncol. 2009, 27, 4733–4740.
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, 699–708.
- Guo, D.; Bell, E.H.; Mischel, P.; Chakravarti, A. Targeting SREBP-1-driven lipid metabolism to treat cancer. Curr. Pharm. Des. 2014, 20, 2619–2626.
- Bhagavan, N.V.; Ha, C.-E. Chapter 16—Lipids I: Fatty Acids and Eicosanoids. In Essentials of Medical Biochemistry, 2nd ed.; Bhagavan, N.V., Ha, C.-E., Eds.; Academic Press: San Diego, CA, USA, 2015; pp. 269–297.
- Mashimo, T.; Pichumani, K.; Vemireddy, V.; Hatanpaa Kimmo, J.; Singh Dinesh, K.; Sirasanagandla, S.; Nannepaga, S.; Piccirillo Sara, G.; Kovacs, Z.; Foong, C.; et al. Acetate Is a Bioenergetic Substrate for Human Glioblastoma and Brain Metastases. Cell 2014, 159, 1603–1614.
- Liu, M.; Liu, N.; Wang, J.; Fu, S.; Wang, X.; Chen, D. Acetyl-CoA Synthetase 2 as a Therapeutic Target in Tumor Metabolism. Cancers 2022, 14, 2896.
- Etchegaray, J.P.; Mostoslavsky, R. Interplay between Metabolism and Epigenetics: A Nuclear Adaptation to Environmental Changes. Mol. Cell 2016, 62, 695–711.
- Rohrig, F.; Schulze, A. The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 2016, 16, 732–749.
- Shen, H.; Shi, L.Z. Metabolic regulation of TH17 cells. Mol. Immunol. 2019, 109, 81–87.
- Cheng, C.; Ru, P.; Geng, F.; Liu, J.; Yoo, J.Y.; Wu, X.; Cheng, X.; Euthine, V.; Hu, P.; Guo, J.Y.; et al. Glucose-Mediated N-glycosylation of SCAP Is Essential for SREBP-1 Activation and Tumor Growth. Cancer Cell 2015, 28, 569–581.
- Horton, J.D. Sterol regulatory element-binding proteins: Transcriptional activators of lipid synthesis. Biochem. Soc. Trans. 2002, 30 Pt 6, 1091–1095.
- Melone, M.A.B.; Valentino, A.; Margarucci, S.; Galderisi, U.; Giordano, A.; Peluso, G. The carnitine system and cancer metabolic plasticity. Cell Death Dis. 2018, 9, 228.
- Schlaepfer, I.R.; Joshi, M. CPT1A-mediated Fat Oxidation, Mechanisms, and Therapeutic Potential. Endocrinology 2020, 161, bqz046.
- Wen, Y.-A.; Xiong, X.; Zaytseva, Y.Y.; Napier, D.L.; Vallee, E.; Li, A.T.; Wang, C.; Weiss, H.L.; Evers, B.M.; Gao, T. Downregulation of SREBP inhibits tumor growth and initiation by altering cellular metabolism in colon cancer. Cell Death Dis. 2018, 9, 265.
- Jie, Z.; Xie, Z.; Xu, W.; Zhao, X.; Jin, G.; Sun, X.; Huang, B.; Tang, P.; Wang, G.; Shen, S.; et al. SREBP-2 aggravates breast cancer associated osteolysis by promoting osteoclastogenesis and breast cancer metastasis. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 115–125.
- Yao, L.; Chen, S.; Li, W. Fatostatin inhibits the development of endometrial carcinoma in endometrial carcinoma cells and a xenograft model by targeting lipid metabolism. Arch. Bio-Chem. Biophys. 2020, 684, 108327.
- Brovkovych, V.; Izhar, Y.; Danes, J.M.; Dubrovskyi, O.; Sakallioglu, I.T.; Morrow, L.M.; Atilla-Gokcumen, G.E.; Frasor, J. Fatostatin induces pro- and anti-apoptotic lipid accumulation in breast cancer. Oncogenesis 2018, 7, 66.
- Li, X.; Chen, Y.T.; Hu, P.; Huang, W.C. Fatostatin displays high antitumor activity in prostate cancer by blocking SREBP-regulated metabolic pathways and androgen receptor signaling. Mol. Cancer Ther. 2014, 13, 855–866.
- Yin, F.; Feng, F.; Wang, L.; Wang, X.; Li, Z.; Cao, Y. SREBP-1 inhibitor Betulin enhances the antitumor effect of Sorafenib on hepatocellular carcinoma via restricting cellular glycolytic activity. Cell Death Dis. 2019, 10, 672.
- Li, J.; Yan, H.; Zhao, L.; Jia, W.; Yang, H.; Liu, L.; Zhou, X.; Miao, P.; Sun, X.; Song, S.; et al. Inhibition of SREBP increases gefitinib sensitivity in non-small cell lung cancer cells. Oncotarget 2016, 7, 52392–52403.
- Caruana, B.T.; Skoric, A.; Brown, A.J.; Lutze-Mann, L.H. Site-1 protease, a novel metabolic target for glioblastoma. Biochem. Biophys. Res. Commun. 2017, 490, 760–766.
- Wang, T.-B.; Geng, M.; Jin, H.; Tang, A.-G.; Sun, H.; Zhou, L.-Z.; Chen, B.-H.; Shen, G.; Sun, Q. SREBP1 site 1 protease inhibitor PF-429242 suppresses renal cell carcinoma cell growth. Cell Death Dis. 2021, 12, 717.
- Siqingaowa Sekar, S.; Gopalakrishnan, V.; Taghibiglou, C. Sterol regulatory element-binding protein 1 inhibitors decrease pancreatic cancer cell viability and proliferation. Biochem. Biophys. Res. Commun. 2017, 488, 136–140.
- Esquejo, R.M.; Roqueta-Rivera, M.; Shao, W.; Phelan, P.E.; Seneviratne, U.; Am Ende, C.W.; Hershberger, P.M.; Machamer, C.E.; Espenshade, P.J.; Osborne, T.F. Dipyridamole Inhibits Lipogenic Gene Expression by Retaining SCAP-SREBP in the Endoplasmic Reticulum. Cell Chem. Biol. 2021, 28, 169–179.e7.
- Granchi, C. ATP-citrate lyase (ACLY) inhibitors as therapeutic agents: A patenting perspective. Expert Opin. Ther. Pat. 2022, 32, 731–742.
- Hatzivassiliou, G.; Zhao, F.; Bauer, D.E.; Andreadis, C.; Shaw, A.N.; Dhanak, D.; Hingorani, S.R.; Tuveson, D.A.; Thompson, C.B. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell 2005, 8, 311–321.
- Corbet, C.; Bastien, E.; Santiago de Jesus, J.P.; Dierge, E.; Martherus, R.; Vander Linden, C.; Doix, B.; Degavre, C.; Guilbaud, C.; Petit, L.; et al. TGFβ2-induced formation of lipid droplets supports acidosis-driven EMT and the metastatic spreading of cancer cells. Nat. Commun. 2020, 11, 454.
- Svensson, R.U.; Parker, S.J.; Eichner, L.J.; Kolar, M.J.; Wallace, M.; Brun, S.N.; Lombardo, P.S.; Van Nostrand, J.L.; Hutchins, A.; Vera, L.; et al. Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small-cell lung cancer in preclinical models. Nat. Med. 2016, 22, 1108–1119.
- Lally, J.S.V.; Ghoshal, S.; DePeralta, D.K.; Moaven, O.; Wei, L.; Masia, R.; Erstad, D.J.; Fujiwara, N.; Leong, V.; Houde, V.P.; et al. Inhibition of Acetyl-CoA Carboxylase by Phosphorylation or the Inhibitor ND-654 Suppresses Lipogenesis and Hepatocellular Carcinoma. Cell Metab. 2019, 29, 174–182.e175.
- Tan, K. Investigating the Role of Acetyl-CoA Carboxylases in Breast Cancer Progression. Doctoral Dissertation, The University of Sydney, Camperdown, Australia, 2021.
- Pizer, E.S.; Thupari, J.; Han, W.F.; Pinn, M.L.; Chrest, F.J.; Frehywot, G.L.; Townsend, C.A.; Kuhajda, F.P. Malonyl-coenzyme-A is a potential mediator of cytotoxicity induced by fatty-acid synthase inhibition in human breast cancer cells and xenografts. Cancer Res. 2000, 60, 213–218.
- Qi, J.; Lang, W.; Geisler, J.G.; Wang, P.; Petrounia, I.; Mai, S.; Smith, C.; Askari, H.; Struble, G.T.; Williams, R.; et al. The use of stable isotope-labeled glycerol and oleic acid to differentiate the hepatic functions of DGAT1 and-2. J. Lipid Res. 2012, 53, 1106–1116.
- Balaban, S.; Nassar, Z.D.; Zhang, A.Y.; Hosseini-Beheshti, E.; Centenera, M.M.; Schreuder, M.; Lin, H.-M.; Aishah, A.; Varney, B.; Liu-Fu, F.; et al. Extracellular fatty acids are the major contributor to lipid synthesis in prostate cancer. Mol. Cancer Res. 2019, 17, 949–962.
- Dow, R.L.; Li, J.C.; Pence, M.P.; Gibbs, E.M.; LaPerle, J.L.; Litchfield, J.; Piotrowski, D.W.; Munchhof, M.J.; Manion, T.B.; Zavadoski, W.J.; et al. Discovery of PF-04620110, a Potent, Selective, and Orally Bioavailable Inhibitor of DGAT-1. ACS Med. Chem. Lett. 2011, 2, 407–412.
- Ren, M.; Xu, H.; Xia, H.; Tang, Q.; Bi, F. Simultaneously targeting SOAT1 and CPT1A ameliorates hepatocellular carcinoma by disrupting lipid homeostasis. Cell Death Discov. 2021, 7, 125.
- Zhu, Y.; Gu, L.; Lin, X.; Zhou, X.; Lu, B.; Liu, C.; Li, Y.; Prochownik, E.V.; Karin, M.; Wang, F.; et al. P53 deficiency affects cholesterol esterification to exacerbate hepatocarcinogenesis. Hepatology 2022.
- Smith, D.C.; Kroiss, M.; Kebebew, E.; Habra, M.A.; Chugh, R.; Schneider, B.J.; Fassnacht, M.; Jafarinasabian, P.; Ijzerman, M.M.; Lin, V.H.; et al. A phase 1 study of nevanimibe HCl, a novel adrenal-specific sterol O-acyltransferase 1 (SOAT1) inhibitor, in adrenocortical carcinoma. Investig. New Drugs 2020, 38, 1421–1429.
- Falchook, G.; Infante, J.; Arkenau, H.T.; Patel, M.R.; Dean, E.; Borazanci, E.; Brenner, A.; Cook, N.; Lopez, J.; Pant, S.; et al. First-in-human study of the safety, pharmacokinetics, and pharmacodynamics of first-in-class fatty acid synthase inhibitor TVB-2640 alone and with a taxane in advanced tumors. EClinicalMedicine 2021, 34, 100797.
- Zhu, Y.; Gu, L.; Lin, X.; Zhang, J.; Tang, Y.; Zhou, X.; Lu, B.; Lin, X.; Liu, C.; Prochownik, E.V.; et al. Ceramide-mediated gut dysbiosis enhances cholesterol esterification and promotes colorectal tu-morigenesis in mice. JCI Insight 2022, 7, e150607.
- Ohmoto, T.; Nishitsuji, K.; Yoshitani, N.; Mizuguchi, M.; Yanagisawa, Y.; Saito, H.; Sakashita, N. K604, a specific acyl-CoA:cholesterol acyltransferase 1 inhibitor, suppresses proliferation of U251-MG glioblastoma cells. Mol. Med. Rep. 2015, 12, 6037–6042.
- Rae, C.; Fragkoulis, G.I.; Chalmers, A.J. Cytotoxicity and Radiosensitizing Activity of the Fatty Acid Synthase Inhibitor C75 Is Enhanced by Blocking Fatty Acid Uptake in Prostate Cancer Cells. Adv. Radiat. Oncol. 2020, 5, 994–1005.
- Grube, S.; Dünisch, P.; Freitag, D.; Klausnitzer, M.; Sakr, Y.; Walter, J.; Kalff, R.; Ewald, C. Overexpression of fatty acid synthase in human gliomas correlates with the WHO tumor grade and inhibition with Orlistat reduces cell viability and triggers apoptosis. J. Neuro-Oncol. 2014, 118, 277–287.
- Li, J.; Condello, S.; Thomes-Pepin, J.; Ma, X.; Xia, Y.; Hurley, T.D.; Matei, D.; Cheng, J.X. Lipid Desaturation Is a Metabolic Marker and Therapeutic Target of Ovarian Cancer Stem Cells. Cell Stem Cell 2017, 20, 303–314.e5.
- Hu, X.; Xiang, J.; Li, Y.; Xia, Y.; Xu, S.; Gao, X.; Qiao, S. Inhibition of Stearoyl-CoA Desaturase 1 Potentiates Anti-tumor Activity of Amodiaquine in Non-small Cell Lung Cancer. Biol. Pharm. Bull. 2022, 45, 438–445.
- Skrypek, K.; Balog, S.; Eriguchi, Y.; Asahina, K. Inhibition of Stearoyl-CoA Desaturase Induces the Unfolded Protein Response in Pancreatic Tumors and Suppresses Their Growth. Pancreas 2021, 50, 219–226.
- She, K.; Fang, S.; Du, W.; Fan, X.; He, J.; Pan, H.; Huang, L.; He, P.; Huang, J. SCD1 is required for EGFR-targeting cancer therapy of lung cancer via re-activation of EGFR/PI3K/AKT signals. Cancer Cell Int. 2019, 19, 103.
- von Roemeling, C.A.; Marlow, L.A.; Wei, J.J.; Cooper, S.J.; Caulfield, T.R.; Wu, K.; Tan, W.W.; Tun, H.W.; Copland, J.A. Stearoyl-CoA desaturase 1 is a novel molecular therapeutic target for clear cell renal cell carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 2368–2380.
- Pisanu, M.E.; Noto, A.; De Vitis, C.; Morrone, S.; Scognamiglio, G.; Botti, G.; Venuta, F.; Diso, D.; Jakopin, Z.; Padula, F.; et al. Blockade of Stearoyl-CoA-desaturase 1 activity reverts resistance to cisplatin in lung cancer stem cells. Cancer Lett. 2017, 406, 93–104.
- Pisanu, M.E.; Maugeri-Saccà, M.; Fattore, L.; Bruschini, S.; De Vitis, C.; Tabbì, E.; Bellei, B.; Migliano, E.; Kovacs, D.; Camera, E.; et al. Inhibition of Stearoyl-CoA desaturase 1 reverts BRAF and MEK inhibition-induced selection of cancer stem cells in BRAF-mutated melanoma. J. Exp. Clin. Cancer Res. CR 2018, 37, 318.
- Oballa, R.M.; Belair, L.; Black, W.C.; Bleasby, K.; Chan, C.C.; Desroches, C.; Du, X.; Gordon, R.; Guay, J.; Guiral, S.; et al. Development of a liver-targeted stearoyl-CoA desaturase (SCD) inhibitor (MK-8245) to establish a therapeutic window for the treatment of diabetes and dyslipidemia. J. Med. Chem. 2011, 54, 5082–5096.
- Sabnis, R.W. Novel Substituted Tetrazoles as ACSS2 Inhibitors for Treating Cancer. ACS Med. Chem. Lett. 2021, 12, 1894–1895.
- Sabnis, R.W. Amide-Substituted Condensed Pyridine Derivatives as ACSS2 Inhibitors for Treating Cancer. ACS Med. Chem. Lett. 2021, 12, 1870–1871.
- Kalthoff, C.; Xiang, K.; Muench, C.; Hlouschek, J.; Jendrossek, V.; Matschke, J. Abstract PO-022: Inhibition of the citrate carrier SLC25A1 affects repair of radiation-induced DNA damage, increases radiosensitivity, and enhances lethality of IR in combination with DNA repair defects or DSB inhibitors. Clin. Cancer Res. 2021, 27 (Suppl. 8), PO-022.
- Hlouschek, J.; Hansel, C.; Jendrossek, V.; Matschke, J. The Mitochondrial Citrate Carrier (SLC25A1) Sustains Redox Homeostasis and Mitochondrial Metabolism Supporting Radioresistance of Cancer Cells With Tolerance to Cycling Severe Hypoxia. Front. Oncol. 2018, 8, 170.
- Reis, L.M.D.; Adamoski, D.; Ornitz Oliveira Souza, R.; Rodrigues Ascenção, C.F.; Sousa de Oliveira, K.R.; Corrêa-da-Silva, F.; Malta de Sá Patroni, F.; Meira Dias, M.; Consonni, S.R.; Mendes de Moraes-Vieira, P.M.; et al. Dual inhibition of glutaminase and carnitine palmitoyltransferase decreases growth and migration of glutaminase inhibition-resistant triple-negative breast cancer cells. J. Biol. Chem. 2019, 294, 9342–9357.
- Jiang, N.; Xie, B.; Xiao, W.; Fan, M.; Xu, S.; Duan, Y.; Hamsafar, Y.; Evans, A.C.; Huang, J.; Zhou, W.; et al. Fatty acid oxidation fuels glioblastoma radioresistance with CD47-mediated immune evasion. Nat. Commun. 2022, 13, 1511.
- Galicia-Vázquez, G.; Aloyz, R. Ibrutinib Resistance Is Reduced by an Inhibitor of Fatty Acid Oxidation in Primary CLL Lymphocytes. Front. Oncol. 2018, 8, 411.
- Flaig, T.W.; Salzmann-Sullivan, M.; Su, L.J.; Zhang, Z.; Joshi, M.; Gijón, M.A.; Kim, J.; Arcaroli, J.J.; Van Bokhoven, A.; Lucia, M.S.; et al. Lipid catabolism inhibition sensitizes prostate cancer cells to antiandrogen blockade. Oncotarget 2017, 8, 56051–56065.
- Schlaepfer, I.R.; Rider, L.; Rodrigues, L.U.; Gijón, M.A.; Pac, C.T.; Romero, L.; Cimic, A.; Sirintrapun, S.J.; Glodé, L.M.; Eckel, R.H.; et al. Lipid catabolism via CPT1 as a therapeutic target for prostate cancer. Mol. Cancer Ther. 2014, 13, 2361–2371.
- Dhakal, B.; Li, C.M.Y.; Li, R.; Yeo, K.; Wright, J.A.; Gieniec, K.A.; Vrbanac, L.; Sammour, T.; Lawrence, M.; Thomas, M.; et al. The Antianginal Drug Perhexiline Displays Cytotoxicity against Colorectal Cancer Cells In Vitro: A Potential for Drug Repurposing. Cancers 2022, 14, 1043.
- Kant, S.; Kesarwani, P.; Guastella, A.R.; Kumar, P.; Graham, S.F.; Buelow, K.L.; Nakano, I.; Chinnaiyan, P. Perhexiline Demonstrates FYN-mediated Antitumor Activity in Glioblastoma. Mol. Cancer Ther. 2020, 19, 1415–1422.
- Ricciardi, M.R.; Mirabilii, S.; Allegretti, M.; Licchetta, R.; Calarco, A.; Torrisi, M.R.; Foà, R.; Nicolai, R.; Peluso, G.; Tafuri, A. Targeting the leukemia cell metabolism by the CPT1a inhibition: Functional preclinical effects in leukemias. Blood 2015, 126, 1925–1929.
- Zhou, J.; Simon, J.M.; Liao, C.; Zhang, C.; Hu, L.; Zurlo, G.; Liu, X.; Fan, C.; Hepperla, A.; Jia, L.; et al. An oncogenic JMJD6-DGAT1 axis tunes the epigenetic regulation of lipid droplet formation in clear cell renal cell carcinoma. Mol. Cell, 2022; in press.
- Martinez-Reyes, I.; Chandel, N.S. Cancer metabolism: Looking forward. Nat. Rev. Cancer 2021, 21, 669–680.
- Zhou, W.; Wahl, D.R. Metabolic Abnormalities in Glioblastoma and Metabolic Strategies to Overcome Treatment Resistance. Cancers 2019, 11, 1231.
- Williams, K.J.; Argus, J.P.; Zhu, Y.; Wilks, M.Q.; Marbois, B.N.; York, A.G.; Kidani, Y.; Pourzia, A.L.; Akhavan, D.; Lisiero, D.N.; et al. An essential requirement for the SCAP/SREBP signaling axis to protect cancer cells from lipotoxicity. Cancer Res. 2013, 73, 2850–2862.
- Ru, P.; Hu, P.; Geng, F.; Mo, X.; Cheng, C.; Yoo, J.Y.; Cheng, X.; Wu, X.; Guo, J.Y.; Nakano, I.; et al. Feedback Loop Regulation of SCAP/SREBP-1 by miR-29 Modulates EGFR Signaling-Driven Glioblastoma Growth. Cell Rep. 2016, 16, 1527–1535.
- Ru, P.; Guo, D. microRNA-29 mediates a novel negative feedback loop to regulate SCAP/SREBP-1 and lipid metabolism. RNA Dis. 2017, 4, e1525.
- Venneti, S.; Dunphy, M.P.; Zhang, H.; Pitter, K.L.; Zanzonico, P.; Campos, C.; Carlin, S.D.; La Rocca, G.; Lyashchenko, S.; Ploessl, K.; et al. Glutamine-based PET imaging facilitates enhanced metabolic evaluation of gliomas in vivo. Sci. Transl. Med. 2015, 7, 274ra217.
- Cheng, C.; Geng, F.; Li, Z.; Zhong, Y.; Wang, H.; Cheng, X.; Zhao, Y.; Mo, X.; Horbinski, C.; Duan, W.; et al. Ammonia stimulates SCAP/Insig dissociation and SREBP-1 activation to promote lipogenesis and tumour growth. Nat. Metab. 2022, 4, 575–588.
- Goldstein, J.L.; DeBose-Boyd, R.A.; Brown, M.S. Protein sensors for membrane sterols. Cell 2006, 124, 35–46.
- Goldstein, J.L.; Brown, M.S. A century of cholesterol and coronaries: From plaques to genes to statins. Cell 2015, 161, 161–172.
- Jeon, T.I.; Osborne, T.F. SREBPs: Metabolic integrators in physiology and metabolism. Trends Endocrinol. Metab. 2012, 23, 65–72.
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131.
- Brown, M.S.; Radhakrishnan, A.; Goldstein, J.L. Retrospective on Cholesterol Homeostasis: The Central Role of Scap. Annu. Rev. Biochem. 2018, 87, 783–807.
- Bennett, M.K.; Lopez, J.M.; Sanchez, H.B.; Osborne, T.F. Sterol regulation of fatty acid synthase promoter. Coordinate feedback regulation of two major lipid pathways. J. Biol. Chem. 1995, 270, 25578–25583.
- Sun, L.P.; Seemann, J.; Goldstein, J.L.; Brown, M.S. Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: Insig renders sorting signal in Scap inaccessible to COPII proteins. Proc. Natl Acad. Sci. USA 2007, 104, 6519–6526.
- Espenshade, P.J.; Cheng, D.; Goldstein, J.L.; Brown, M.S. Autocatalytic processing of site-1 protease removes propeptide and permits cleavage of sterol regulatory element-binding proteins. J. Biol. Chem. 1999, 274, 22795–22804.
- Cheng, D.; Espenshade, P.J.; Slaughter, C.A.; Jaen, J.C.; Brown, M.S.; Goldstein, J.L. Secreted site-1 protease cleaves peptides corresponding to luminal loop of sterol regulatory element-binding proteins. J. Biol. Chem. 1999, 274, 22805–22812.
- Rawson, R.B.; Zelenski, N.G.; Nijhawan, D.; Ye, J.; Sakai, J.; Hasan, M.T.; Chang, T.Y.; Brown, M.S.; Goldstein, J.L. Complementation cloning of S2P, a gene encoding a putative metalloprotease required for intramembrane cleavage of SREBPs. Mol. Cell 1997, 1, 47–57.
- Duncan, E.A.; Dave, U.P.; Sakai, J.; Goldstein, J.L.; Brown, M.S. Second-site cleavage in sterol regulatory element-binding protein occurs at transmembrane junction as determined by cysteine panning. J. Biol. Chem. 1998, 273, 17801–17809.
- Hua, X.; Sakai, J.; Brown, M.S.; Goldstein, J.L. Regulated cleavage of sterol regulatory element binding proteins requires sequences on both sides of the endoplasmic reticulum membrane. J. Biol. Chem. 1996, 271, 10379–10384.
- Yang, T.; Espenshade, P.J.; Wright, M.E.; Yabe, D.; Gong, Y.; Aebersold, R.; Goldstein, J.L.; Brown, M.S. Crucial step in cholesterol homeostasis: Sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell 2002, 110, 489–500.
- Yabe, D.; Brown, M.S.; Goldstein, J.L. Insig-2, a second endoplasmic reticulum protein that binds SCAP and blocks export of sterol regulatory element-binding proteins. Proc. Natl. Acad. Sci. USA 2002, 99, 12753–12758.
- Espenshade, P.J.; Li, W.P.; Yabe, D. Sterols block binding of COPII proteins to SCAP, thereby controlling SCAP sorting in ER. Proc. Natl. Acad. Sci. USA 2002, 99, 11694–11699.
- Adams, C.M.; Goldstein, J.L.; Brown, M.S. Cholesterol-induced conformational change in SCAP enhanced by Insig proteins and mimicked by cationic amphiphiles. Proc. Natl. Acad. Sci. USA 2003, 100, 10647–10652.
- Guo, D. SCAP links glucose to lipid metabolism in cancer cells. Mol. Cell. Oncol. 2016, 3, e1132120.
- Shao, W.; Espenshade, P.J. Sugar Makes Fat by Talking to SCAP. Cancer Cell 2015, 28, 548–549.
- Cheng, C.; Guo, J.Y.; Geng, F.; Wu, X.; Cheng, X.; Li, Q.; Guo, D. Analysis of SCAP N-glycosylation and Trafficking in Human Cells. J. Vis. Exp. 2016, 117, e54709.
- Yang, Y.; Hu, B. 21-Bio-based chemicals from biorefining: Lipid and wax conversion and utilization. In Advances in Biorefineries; Waldron, K., Ed.; Woodhead Publishing: Thorston, UK, 2014; pp. 693–720.
- Currie, E.; Schulze, A.; Zechner, R.; Walther Tobias, C.; Farese Robert, V. Cellular Fatty Acid Metabolism and Cancer. Cell Metab. 2013, 18, 153–161.
- Hoy, A.J.; Nagarajan, S.R.; Butler, L.M. Tumour fatty acid metabolism in the context of therapy resistance and obesity. Nat. Rev. Cancer 2021, 21, 753–766.
- Pérez-Chacón, G.; Astudillo, A.M.; Balgoma, D.; Balboa, M.A.; Balsinde, J. Control of free arachidonic acid levels by phospholipases A2 and lysophospholipid acyltransferases. Biochim. Bi-Ophysica Acta (BBA)-Mol. Cell Biol. Lipids 2009, 1791, 1103–1113.
- Cifarelli, V.; Abumrad, N.A. Chapter 48—Enterocyte Fatty Acid Handling Proteins and Chylomicron Formation. In Physiology of the Gastrointestinal Tract, 6th ed.; Said, H.M., Ed.; Academic Press: Cambridge, MA, USA, 2018; pp. 1087–1107.
- Wang, B.; Tontonoz, P. Phospholipid Remodeling in Physiology and Disease. Annu. Rev. Physiol. 2019, 81, 165–188.
- Saito, R.F.; Rangel, M.C.; Halman, J.R.; Chandler, M.; de Sousa Andrade, L.N.; Odete-Bustos, S.; Furuya, T.K.; Carrasco, A.G.M.; Chaves-Filho, A.B.; Yoshinaga, M.Y.; et al. Simultaneous silencing of lyso-phosphatidylcholine acyltransferases 1–4 by nucleic acid nanoparticles (NANPs) improves radiation response of melanoma cells. Nanomed. Nanotechnol. Biol. Med. 2021, 36, 102418.
- Shi, C.; Qiao, S.; Wang, S.; Wu, T.; Ji, G.J.I.J.C.E.M. Recent progress of lysophosphatidylcholine acyltransferases in metabolic disease and cancer. Int. J. Clin. Exp. Med. 2018, 11, 8941–8953.
- Bi, J.; Ichu, T.-A.; Zanca, C.; Yang, H.; Zhang, W.; Gu, Y.; Chowdhry, S.; Reed, A.; Ikegami, S.; Turner, K.M.; et al. Oncogene Amplification in Growth Factor Signaling Pathways Renders Cancers De-pendent on Membrane Lipid Remodeling. Cell Metab. 2019, 30, 525–538.e8.
- He, R.-Q.; Li, J.-D.; Du, X.-F.; Dang, Y.-W.; Yang, L.-J.; Huang, Z.-G.; Liu, L.-M.; Liao, L.-F.; Yang, H.; Chen, G. LPCAT1 overexpression promotes the progression of hepatocellular carcinoma. Cancer Cell Int. 2021, 21, 442.
- Du, Y.; Wang, Q.; Zhang, X.; Wang, X.; Qin, C.; Sheng, Z.; Yin, H.; Jiang, C.; Li, J.; Xu, T. Lysophosphatidylcholine acyltransferase 1 upregulation and concomitant phospholipid alterations in clear cell renal cell carcinoma. J. Exp. Clin. Cancer Res. CR 2017, 36, 66.
- Tao, M.; Luo, J.; Gu, T.; Yu, X.; Song, Z.; Jun, Y.; Gu, H.; Han, K.; Huang, X.; Yu, W.; et al. LPCAT1 reprogramming cholesterol metabolism promotes the progression of esophageal squamous cell car-cinoma. Cell Death Dis. 2021, 12, 845.
- Uehara, T.; Kikuchi, H.; Miyazaki, S.; Iino, I.; Setoguchi, T.; Hiramatsu, Y.; Ohta, M.; Kamiya, K.; Morita, Y.; Tanaka, H.; et al. Overexpression of Lysophosphatidylcholine Acyltransferase 1 and Con-comitant Lipid Alterations in Gastric Cancer. Ann. Surg. Oncol. 2016, 23 (Suppl. 2), S206–S213.
- Lebok, P.; von Hassel, A.; Meiners, J.; Hube-Magg, C.; Simon, R.; Höflmayer, D.; Hinsch, A.; Dum, D.; Fraune, C.; Göbel, C.; et al. Up-regulation of lysophosphatidylcholine acyltransferase 1 (LPCAT1) is linked to poor prognosis in breast cancer. Aging 2019, 11, 7796–7804.
- Li, D.; Li, Y. The interaction between ferroptosis and lipid metabolism in cancer. Signal Transduct. Target. Ther. 2020, 5, 108.
- Mbah, N.E.; Lyssiotis, C.A. Metabolic regulation of ferroptosis in the tumor microenvironment. J. Biol. Chem. 2022, 298, 101617.
- Lei, P.; Bai, T.; Sun, Y. Mechanisms of Ferroptosis and Relations with Regulated Cell Death: A Review. Front. Physiol. 2019, 10, 139.
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90.
- Yeagle, P. L: Modulation of membrane function by cholesterol. Biochimie 1991, 73, 1303–1310.
- Luo, J.; Yang, H.; Song, B.L. Mechanisms and regulation of cholesterol homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 225–245.
- Holthuis, J.C.; Menon, A.K. Lipid landscapes and pipelines in membrane homeostasis. Nature 2014, 510, 48–57.
- Ikonen, E. Cellular cholesterol trafficking and compartmentalization. Nat. Rev. Mol. Cell Biol. 2008, 9, 125–138.
- van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124.
- Iaea, D.B.; Maxfield, F.R. Cholesterol trafficking and distribution. Essays Biochem. 2015, 57, 43–55.
- Holmes, M.V.; Ala-Korpela, M. What is ‘LDL cholesterol’? Nat. Rev. Cardiol. 2019, 16, 197–198.
- Maxfield, F.R.; van Meer, G. Cholesterol, the central lipid of mammalian cells. Curr. Opin. Cell Biol. 2010, 22, 422–429.
- Segrest, J.P.; Jones, M.K.; De Loof, H.; Dashti, N. Structure of apolipoprotein B-100 in low density lipoproteins. J. Lipid Res. 2001, 42, 1346–1367.
- Goldstein, J.L.; Brown, M.S. The LDL receptor. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 431–438.
- Goldstein, J.L.; Anderson, R.G.; Brown, M.S. Receptor-mediated endocytosis and the cellular uptake of low density lipoprotein. Ciba Found. Symp. 1982, 92, 77–95.
- Goldstein, J.L.; Basu, S.K.; Brown, M.S. Receptor-mediated endocytosis of low-density lipoprotein in cultured cells. Methods Enzymol. 1983, 98, 241–260.
- Ertunc, M.E.; Hotamisligil, G.S. Lipid signaling and lipotoxicity in metaflammation: Indications for metabolic disease pathogenesis and treatment. J. Lipid Res. 2016, 57, 2099–2114.
- Lipke, K.; Kubis-Kubiak, A.; Piwowar, A. Molecular Mechanism of Lipotoxicity as an Interesting Aspect in the Development of Pathological States—Current View of Knowledge. Cells 2022, 11, 844.
- Olzmann, J.A.; Carvalho, P. Dynamics and functions of lipid droplets. Nat. Rev. Mol. Cell Biol. 2019, 20, 137–155.