2. Standard GBM Therapy
GBM is a WHO grade IV glioma, with a median survival post-diagnosis of only approximately 15 months despite extensive treatments
[23]. GBM represents more than 60% of all brain tumors in adults and occurs in 2–3 cases per 100,000 individuals in Europe and North America
[24]. Approximately 90% of GBM cases occur as primary tumors that have wild-type isocitrate dehydrogenase (IDH) and less than three months of clinical symptoms before diagnosis, while less than 5% of all cases are secondary tumors with IDH mutations, progressing from WHO grade II/III gliomas and with better prognosis
[12][25][26][27][12,25,26,27]. The standard treatment for GBM is to first perform maximal surgical resection, followed by concomitant radiotherapy and alkylating agent Temozolomide (TMZ) for 6 weeks, and then continuation of TMZ alone every 4 weeks for six cycles of maintenance treatment
[28]. Unfortunately, this treatment is only effective for a few months and almost all GBM tumors unavoidably recur after treatment
[29]. The 5-year survival for patients with GBM is only around 6.8% in the United States
[30].
In the clinic, except for TMZ, only Bevacizumab, a humanized monoclonal antibody against vascular endothelial growth factor (VEGF), has been approved by the FDA for the treatment of recurrent GBM and not for newly diagnosed GBM
[31][32][31,32]. It is effective at reducing symptoms and improving the quality of life only for a short time
[25]. However, Bevacizumab has no combinatory effect with TMZ and radiotherapy
[25]. Moreover, it reduces the uptake of TMZ, fails to increase the overall survival, and has significant side effects such as hypertension, venous thrombosis, and infections
[25].
3. Lipid Metabolism Regulation in GBM
3.1. Lipogenesis
Glucose is the main source for lipid production through the de novo synthesis pathway
[1]. Glucose through glycolysis generates pyruvate that is converted to acetyl-CoA in the mitochondria by the pyruvate dehydrogenase (PDH)
[1][33][1,39]. Condensation of acetyl-CoA with oxaloacetate (OAA) forms citrate, which is then released into the cytosol by the SLC25A1 transporter and converted back to acetyl-CoA by ATP-citrate lyase (ACLY). Acetyl-CoA then serves as a precursor for fatty acid and cholesterol synthesis
[1][34][1,40] (
Figure 1). In addition, acetyl-CoA synthetase 2 (ACSS2) can convert cytosolic acetate to acetyl-CoA for de novo fatty acid and cholesterol synthesis (
Figure 1) to promote tumor growth
[1]. ACSS2 is upregulated in GBM
[35][41] and in hepatocellular carcinoma (HCC), myeloma, prostate, and bladder cancers
[36][42]. Furthermore, acetyl-CoA is also involved in epigenetic regulation as it enters into the nucleus to modify histone proteins by direct acetylation
[37][43].
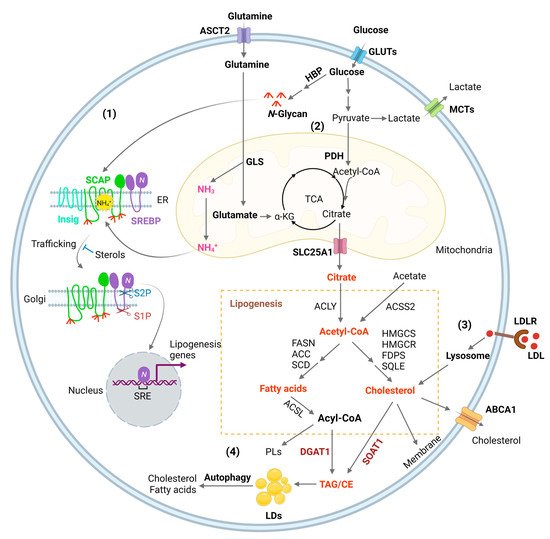
Figure 1. Lipid metabolism reprogramming in cancer cells. (1) Combined elevation of glucose and glutamine consumption promotes lipogenesis by activating the SREBP/SCAP pathway. Glucose produces N-glycans through HBP (Hexosamine Biosynthesis Pathway), which stabilizes SCAP (SREBP-cleavage activating protein) through N-linked glycosylation of its luminal loops (1 and 7). Glutamine enters the mitochondria and ammonia (NH3) is released by GLS (glutaminase). NH3 is protonated and converted to NH4+, which directly binds to the aspartate D428 and Serine S326/330 in the core of the SCAP transmembrane domains, forming stable hydrogen bonds. This binding triggers a dramatic conformation change in SCAP, leading to its dissociation from Insig (insulin-induced gene), an endoplasmic reticulum (ER)-resident protein. Subsequently, SCAP escorts SREBP (sterol regulatory element-binding protein) to the Golgi, where it is cleaved by two enzymes S1P (site 1 protease) and S2P (site 2 protease) to release its active N-terminal fragment. Finally, the N-terminal domain goes into the nucleus, binds to the SRE (sterol regulatory element) motif located in the promoters of gene involved in lipogenesis to activate their transcription and promote de novo lipid synthesis and tumor growth. (2) Glucose is the main source for lipid synthesis. Glucose via glycolysis breaks down into pyruvate, which enters the mitochondria and is converted to acetyl-CoA by PDH (pyruvate dehydrogenase), followed by condensation with OAA (oxaloacetate) to form citrate to enter the TCA cycle (tricarboxylic acid cycle). Citrate is released to the cytosol via its mitochondria transporter, SLC25A1. Citrate is then cleaved to acetyl-CoA by ACLY (ATP citrate lyase), which serves as a precursor for fatty acid and cholesterol biosynthesis catalyzed by a series of enzymes that are the main transcriptional targets of SREBPs, as shown in the Figure. In addition, cytosol acetate can be converted to acetyl-CoA for lipid synthesis by ACCS2 (acetyl-CoA synthetase 2). Besides that, acetyl-CoA is also the substrate for the acetylation of histones, which is involved in epigenetic regulation. Moreover, glutamine through glutaminolysis contributes as an anaplerotic substrate to replenish tricarboxylic acid (TCA) cycle intermediates. Glutamate, the product of the first step of glutaminolysis, is converted to α-KG (α-ketoglutaric acid) and then enters into the TCA cycle. (3) SREBPs upregulates the expression of LDLR (low-density lipoprotein receptor), which binds to LDL and transports it into cells through the endocytosis process. LDL is then hydrolyzed in the lysosomes and cholesterol is released, promoting tumor growth. (4) Excess fatty acids and cholesterol are converted to TAG (triacylglycerol) and CE (cholesteryl ester) by DGAT1 (diacylglycerol O-acyltransferase 1) and sterol O acyl-transferase 1 (SOAT1) to form LDs (lipid droplets) and prevent toxicity from high lipid levels. Under conditions of nutrient deficiency, LDs are hydrolyzed by autophagy to release free fatty acids and cholesterol for tumor survival.
De novo fatty acid synthesis is commonly observed in the liver, adipose tissue, and lactating breast, and normal cells usually assimilate fatty acid through the uptake of extracellular lipids
[38][44]. However, actively proliferating cells, such as cancer cells and effector T cells, increase their fatty acid synthesis, uptake, and oxidation (FAO) for the synthesis of structural lipids and energy expenditure
[9][39][9,45]. Expression of the enzymes controlling de novo fatty acid synthesis, such as ACLY, ACSS2, acetyl-CoA carboxylases (ACC), fatty acid synthase (FASN), and stearoyl-CoA desaturase 1 (SCD1), and mitochondria citrate transporter SLC25A1, is mainly regulated by sterol regulatory element-binding protein 1 (SREBP-1), a master transcription factor that regulates fatty acid and cholesterol synthesis (
Figure 1)
[13][14][15][40][41][13,14,15,46,47]. Inhibitors targeting these enzymes have been developed for the treatment of a broad type of cancers (
Table 1)
[9]. Carnitine palmitoyltransferase 1 (CPT1), the rate-limiting enzyme involved in mitochondrial fatty acid oxidation, which allows fatty acyl-CoA to enter into the mitochondria matrix from the cytosol, is also an important target of lipid metabolism for cancer therapy
[42][43][48,49].
Table 1. Molecular targets and inhibitors in the lipid metabolism pathway for cancer therapy.
| Target |
Inhibitors |
Types of Cancer |
Preclinical Evidence |
Clinical Trials |
References |
| SREBPs |
Fatostatin, Betulin, PF-429242, dipyridamole |
GBM, breast, uterine, prostate, liver, lung,
kidney, pancreatic and colon |
Xenografts |
|
[1][19][44][45][46][47][48][49][50][51][52][53][54] |
| ACLY |
SB-201076, Hydroxycitric acid, Bempedoic acid, BMS-303141, SB-204990, |
Lung, prostate |
Xenografts |
|
[1][55][56] |
| ACCs |
ND-646, ND-654, soraphen A, TOFA |
Lung, liver, breast |
Xenografts,
DEN-injured rats |
|
[17][57][58][59][60][61][62][63][64] |
| FASN |
TVB-2640, C75, cerulenin, orlistat |
Lung, colon, breast, GBM, astrocytoma, prostate, uterine |
Xenografts |
NCT03808558
NCT02223247
NCT02980029
NCT03179904
NCT05118776
NCT03032484 |
[65][66][67][68][69][70][71][72] |
| SCD1 |
A939572, MF-438, CAY10566
MK-8245 |
Lung, pancreatic, kidney, GBM, ovarian |
Xenograft,
Pdx1Cre; LSL-KrasG12D mouse |
NCT00972322
(type 2 diabetes) |
[73][74][75][76][77][78][79][80] |
| ACSS2 |
Tetrazoles, Pyridine Derivatives |
|
|
|
[81][82] |
| SLC25A1 |
CTPi2, BTA, CNASB |
|
|
|
[83][84] |
| CPT1A |
Etomoxir, Perhexiline, ST1326 |
GBM, breast, leukemia, prostate, colon |
Xenografts,
Transgenic mice |
|
[85][86][87][88][89][90][91][92] |
| DGAT1 |
A922500, AZD3988,
PF-04620110 |
GBM, liver, uterine, prostate |
Xenografts |
|
[17][57][62][63][64][93] |
| SOAT1 |
Avasimibe, ATR-101, K604, |
Liver, colon, GBM, kidney |
P53-deficient mice, AOM/DSS-treated and ApcMin/+ mice, xenografts |
NCT01898715 |
[65][66][67][69][70] |
Like in most cancers, metabolism is reprogrammed in GBM and plays an important role during tumor initiation and progression, with increased glycolysis, altered nucleotide metabolism, and glutamine addiction
[94][95][50,51].
ResWe
archers were the first to report that fatty acid synthesis is highly elevated in GBM to promote rapid tumor growth
[1][13][14][15][1,13,14,15].
ResWe
archers found that the oncogenic EGFR/PI3K/AKT pathway increases fatty acid synthesis by stimulating the activation of SREBP-1
[14][33][14,39]. Inactivating SREBP-1 pharmacologically with its inhibitor Fatostatin or by shRNA knockdown significantly suppresses GBM tumor growth in vitro and in vivo
[14][19][96][14,19,52]. Directly targeting FASN, a downstream transcriptional target of SREBP-1 and a key enzyme in controlling de novo fatty acid synthesis, with its inhibitor C75 effectively suppresses GBM growth
[14]. Moreover, inhibiting fatty acid and cholesterol synthesis with the AMP-activated protein kinase (AMPK) agonist AICAR (5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside) also reduces GBM growth in a xenograft mouse model
[13][16][13,16]. Furthermore,
our
esearchers' studies revealed that a small non-coding RNA, miR-29, mediates a negative feedback loop to control SREBP expression and lipogenesis
[97][98][53,54]. SREBP-1 transcriptionally upregulates pre-miR-29 expression, while mature miR-29 can inversely inhibit SREBP-1 expression via binding to the 3′-untranslational region (3-UTR) of SREBP-1 mRNA to induce its degradation and suppress SREBP translation. Delivering miR-29 to GBM cells significantly suppresses tumorigenesis in orthotopic mouse model by inhibiting SREBP-1 and lipogenesis
[97][98][53,54]. Collectively, these data demonstrate that inhibiting lipogenesis is a promising new direction for GBM therapy.
3.2. SREBP Activation, Connecting Glucose and Glutamine to Lipid Synthesis
GBM growth consumes large amounts of glucose and amino acids
[99][56], but the mechanism by which tumor cells sense their levels to trigger lipid synthesis has been left unanswered for a long time.
ReseaOur
chers' most recent study identified that glucose and glutamine coordinate to activate SREBP-1 to trigger de novo lipid synthesis in GBM and various other cancer cells
[100][57]. The SREBP family includes three isoforms, SREBP-1a, -1c, and -2
[101][102][103][58,59,60]. SREBP-1c mainly regulates the expression of genes controlling fatty acid synthesis, while SREBP-2 regulates cholesterol synthesis and uptake, and SREBP-1a, which has the highest transcriptional activity, regulates all three processes
[1][104][105][106][1,61,62,63]. SREBPs are synthesized as ~125 kD inactive precursors, which are spatially restrained in the endoplasmic reticulum (ER) membrane and are activated through a tightly controlled ER–Golgi–nucleus translocation process
[102][105][59,62]. SREBPs bind to SREBP-cleavage activating protein (SCAP), which further binds to COPII-coated vesicles to move from the ER to the Golgi
[105][107][62,64]. In the Golgi, SREBPs are sequentially cleaved by site-1 and -2 proteases to release their N-terminal forms (~65 kD) that then enter the nucleus to activate lipogenic gene expression
[108][109][110][111][112][65,66,67,68,69]. Interestingly, SCAP/SREBP trafficking is inhibited by an ER-resident protein, insulin-inducible gene protein (Insig), which includes two isoforms, Insig-1 and -2
[113][114][70,71]. Insig binds to SCAP to retain the SCAP/SREBP complex in the ER (
Figure 1)
[102][107][59,64]. Cholesterol or 25-hydroxycholesterol (25-HC) can bind to SCAP or Insig to further enhance their association, which represents a negative feedback loop to modulate SREBP activation (
Figure 1)
[113][115][116][70,72,73].
The key step activating the SCAP/Insig dissociation for subsequent SREBP translocation has just been elucidated in a recent study by Cheng et al.
[100][57].
ReseaOur
chers' previous study showed that glucose stimulates SREBP activation and lipogenesis by promoting SCAP N-glycosylation and stability
[40][117][118][119][46,74,75,76]. Unexpectedly,
our
esearchers' most recent study shows that when glutamine is removed from the medium, glucose alone is unable to activate SREBPs and lipogenesis, even with low cholesterol levels and in the presence of SCAP N-glycosylation
[100][57].
RWe
searchers uncovered an unprecedented role of ammonia, which is released by glutaminolysis and acts as a key activator of the dissociation of N-glycosylated SCAP from Insig by inducing dramatic conformational changes in the SCAP transmembrane domain through interaction via hydrogen bonds with the side chains of three residues, i.e., aspartate D428, serine S326 and S330, eventually leading to SREBP activation and lipid synthesis (
Figure 1)
[100][57].
3.3. Membrane Phospholipid Remodeling
Phospholipids (PLs) constitute the main membrane structure as they form lipid bilayers through their amphiphilic characteristics
[120][77]. Elevated fatty acid synthesis promotes PL formation and membrane expansion to facilitate rapid tumor growth
[1][121][122][1,78,79]. The composition of the fatty acid chains in PLs is not static, but is dynamically remodeled through the coordinated activities of phospholipase A (PLA) and lysophosphatidylcholine acyltransferases (LPCATs)
[123][80]. PLA removes an esterified fatty acid from the sn-2 position of PLs to convert them to lysophosphatidylipids, while LPCATs add back fatty acids to lysophosphatidylipids, thereby remaking PLs
[124][125][81,82]. Thus, fatty acid chains in membrane PLs are modified by these two enzymes to adjust to the dynamic alterations of the cellular environment
[125][82]. The LPCAT family contains four members (LPCAT1-4)
[126][127][83,84]. LPCAT1 mainly regulates the incorporation of saturated fatty acids into lysophosphatidylcholine (LPC) to form PLs, thereby increasing the levels of saturated PLs. Interestingly, LPCAT1 has been shown to be upregulated in various cancers, including GBM
[128][85], HCC
[129][86], clear cell renal cell cancer
[130][87], esophageal squamous cell carcinoma (ESCC)
[131][88], gastric
[132][89], and breast cancer
[133][90]. Its upregulation regulates oncogenic EGFR signaling in GBM by affecting EGFR and its constitutively active mutant EGFRvIII protein stability
[128][85]. Targeting LPCAT1 effectively suppresses GBM
[128][85] and ESCC growth
[131][88]. LPCAT1 elevation across cancer types suggests that the increase in membrane saturated PL levels might prevent lipid peroxidation and ferroptosis in cancer cells, thereby facilitating tumor growth. LPCAT3 regulates the incorporation of polyunsaturated fatty acids, such as arachidonic acid, thereby increasing membrane fluidity and flexibility
[127][84]. LPCAT3 has been shown to be a major player to facilitate lipid peroxidation and ferroptosis via increasing membrane polyunsaturated PL level
[134][135][136][137][91,92,93,94]. Nevertheless, there has no study clearly demonstrating the expression levels of LPCPAT3 in tumor tissues and its role in tumorigenesis.
3.4. Cholesterol Uptake
Cholesterol, which is an indispensable lipid molecule for maintaining cell viability
[138][95], is inserted between phospholipid molecules to control membrane integrity and fluidity
[139][96], and accounts for 30–40% of membrane lipids in the plasma membrane
[140][141][142][143][97,98,99,100]. Cholesterol is esterified with fatty acids to convert to neutral cholesteryl esters (CE), which are carried by low-density lipoprotein (LDL) to circulate in the bloodstream and be absorbed by different tissues
[144][101]. LDL is endocytosed into cells as mediated by the LDL receptor (LDLR) and is then delivered to the lysosomes, where LDL is broken down by lysosomal lipases to release free cholesterol, which then egresses from the lysosomes for maintenance and expansion of the plasma membrane but also of the membranes of all cellular organelles such as the mitochondria and endoplasmic reticulum (ER) (
Figure 1)
[15][145][146][147][148][149][15,102,103,104,105,106].
3.5. Lipid storage and Energy Homeostasis
Excess free fatty acids and cholesterol can cause lipotoxicity, leading to cell death when the levels are uncontrolled
[150][151][107,108]. In the human body, excess fatty acids and cholesterol are sequestered into the adipocytes of fat tissues within specialized organelles known as lipid droplets (LDs) after being converted to triacylglycerol (TAG) and cholesteryl esters (CE) (
Figure 1)
[152][109]. Interestingly, in 2016, Geng et al. reported for the first time that LDs are highly prevalent in human GBM tumor tissues, while not observed in normal brain tissues
[19][20][19,20]. Using transmission electronic microscopy, they clearly showed that LDs are present in the cytosol of GBM tumor cells.