Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Conner Chen and Version 3 by Conner Chen.
Hippo signaling pathway is a key modulator of tissue growth with widespread implications in organ development, cell growth, regeneration, and stem cell function.
- Hippo signaling
- TEAD
- transcriptional regulation
1. Introduction
Hippo signaling pathway is a key modulator of tissue growth with widespread implications in organ development, cell growth, regeneration, and stem cell function [1]. It is at the crossroads of several upstream signaling events that control the activation and deactivation of Yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ)—two homologous proteins, collectively known as YAP/TAZ (Drosophila ortholog Yorkie, Yki) (Figure 1). YAP/TAZ transcription complex, when in the nucleus, associates with multiple transcription factors and activates gene networks that signal cell proliferation and survival. Conversely, sequestration of the YAP/TAZ complex in the cytosol is critical to finetune cell fate regulation. YAP/TAZ is phosphorylated by members of the NDR family kinases, Large Tumor Suppressor 1/2 (LATS1/2; Drosophila ortholog Warts, Wts). LATS1/2 is regulated by kinase, Mammalian STE20-like 1/2 (MST1/2; Drosophila homolog Hippo, Hpo), and their adaptor proteins SAV1 (Drosophila homolog Salvador, Sav) and MOB1 (Drosophila homolog Mats) [2]. Phosphorylation of the YAP/TAZ complex by LATS1/2 leads to destabilization of the complex and renders it inaccessible to the nucleus via promoting interaction with cytoplasmic protein 14-3-3. Nuclear localization of YAP/TAZ is critical for its function as transcriptional co-activators [2][3].
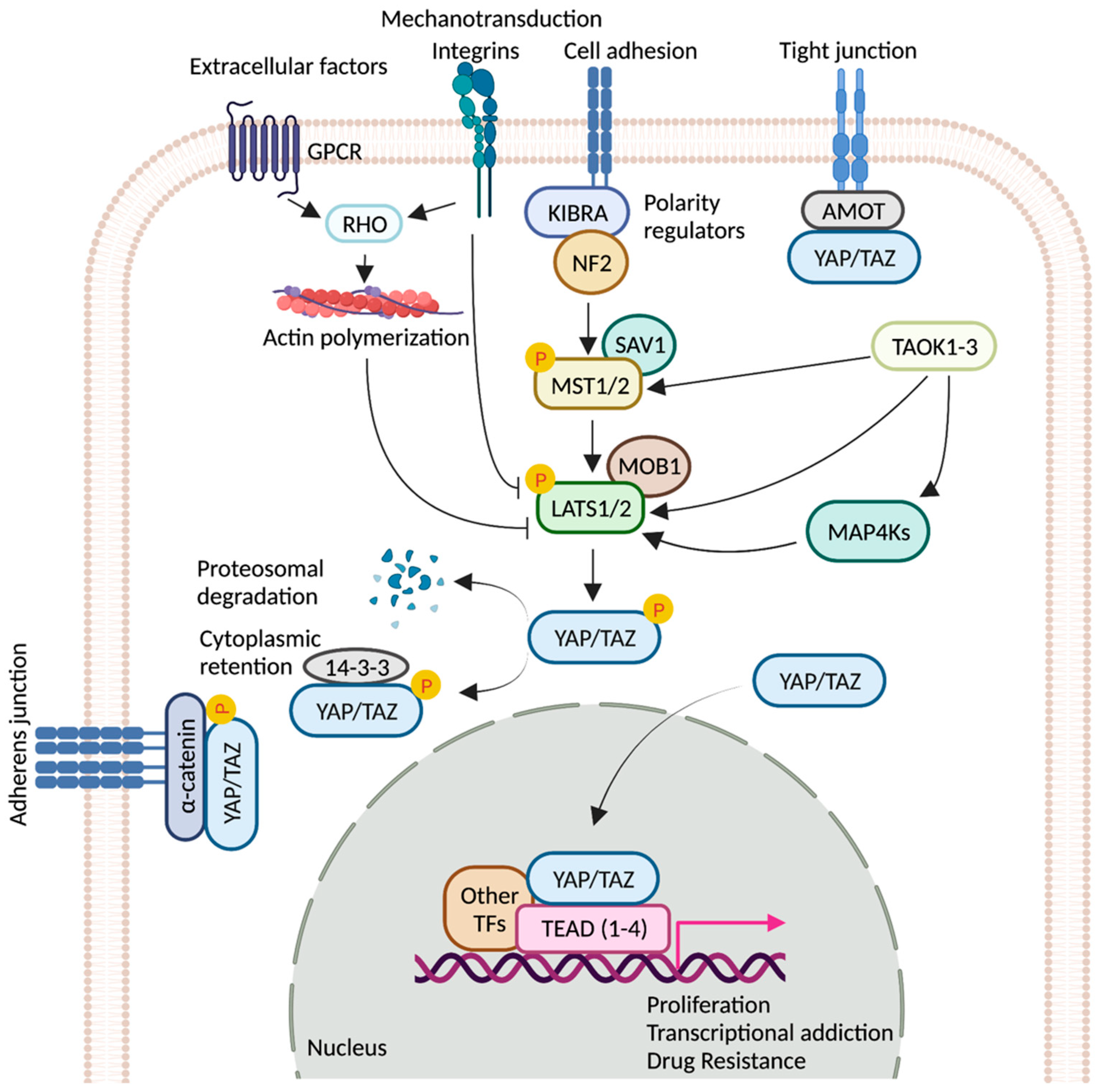
Figure 1. Regulation of YAP/TAZ activity by key signaling events. Schematic representation of the core components of the Hippo pathway. When the pathway is ON, a cascade of core kinases, composed of MST1/2 and LATS1/2, trigger phosphorylation of YAP/TAZ, which results in degradation or cytoplasmic retention of YAP/TAZ by 14-3-3. Various other signaling pathways and upstream effectors such as GPCRs (G protein-coupled receptors), TAOK family kinases, cell polarity, and adhesion regulators influence the activity of YAP/TAZ [4][5][6][7]. Mechanical cues relayed by extracellular-matrix-binding integrins and GPCR-mediated actin polymerization can inactivate the pathway. Unphosphorylated YAP/TAZ translocate into the nucleus, where it interacts with TEAD(1-4) and other cofactors. Together, they fuel the expression of pro-tumorigenic genes that can contribute to metastasis, transcriptional addiction, and drug resistance. Figure created with BioRender.com.
YAP/TAZ lack a DNA-binding domain, and primarily rely on their interaction with additional transcription factors (TFs) to exert transcriptional activity [8][9] (Figure 2). YAP/TAZ can also recruit co-factors and chromatin remodelers, forming distinct transcriptional modules that bind to cis-regulatory elements and govern transcriptional output. This adds layers of regulation to the transcriptional machinery, allowing it to receive information from several upstream signaling events, as well as to finetune gene expression on demand. Conversely, deregulation of YAP/TAZ or its associated factors leads to aberrant gene expression in cancer. YAP/TAZ has also recently emerged as a critical nexus contributing to resistance mechanisms against therapeutic interventions [10][11]. Although most of the YAP-associated gene expression networks require association of the TEAD family of transcription factors (TEAD 1-4) (Drosophila ortholog Scalloped, Sd), TEAD-independent gene regulation networks have also been documented [1].
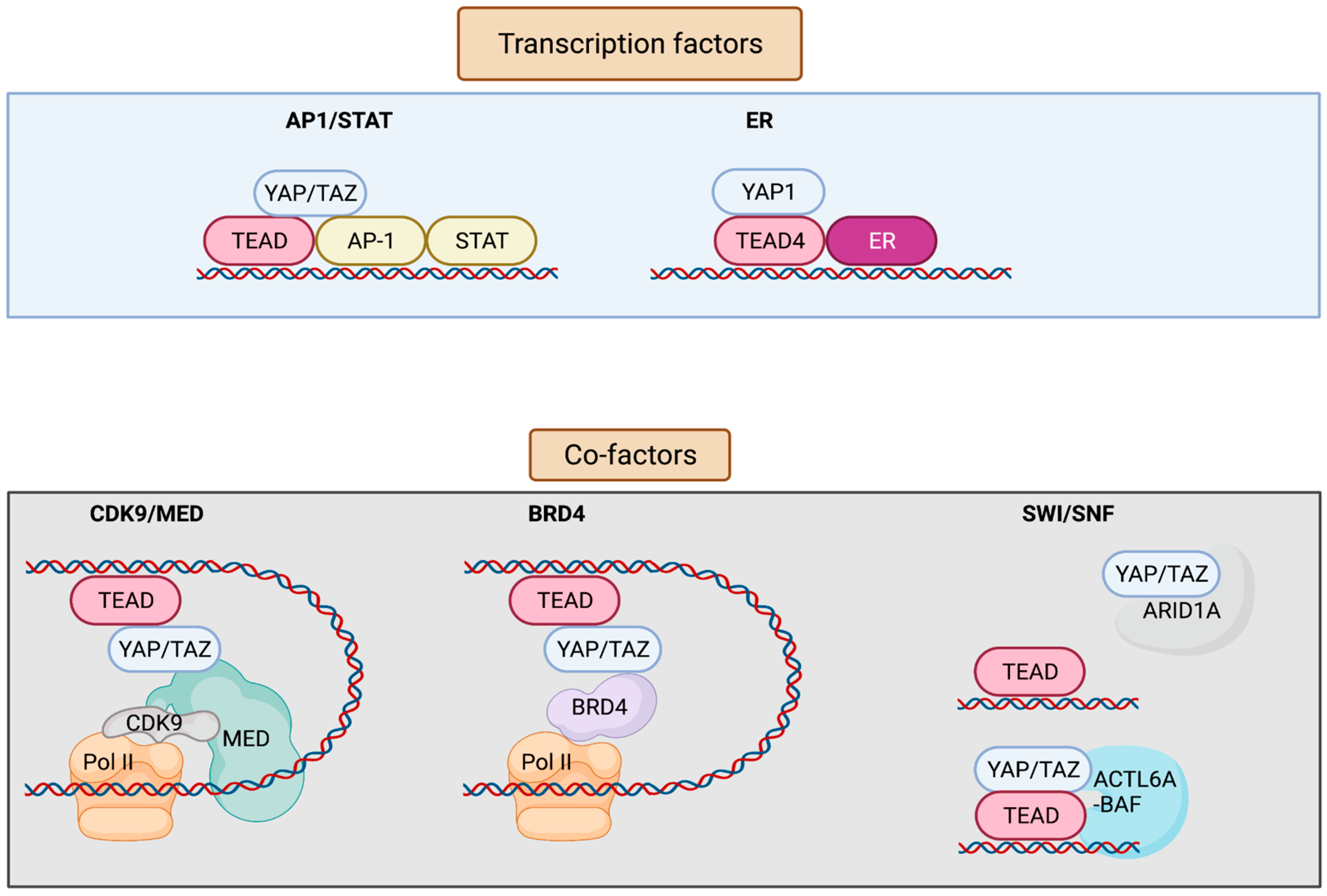
Figure 2. Interaction factors with YAP/TAZ/TEAD. YAP/TAZ/TEAD interact with transcription factors such as AP-1, STATs, and ER to drive transcription. YAP/TAZ/TEAD at enhancers recruit co-factors including Mediator and BRD4 that enable the release of paused Pol II and resumption of transcription elongation. YAP/TAZ/TEAD can also interact with various subunits of the SWI/SNF chromatin remodeling complex. ARID1A is thought to suppress YAP/TAZ transcriptional activity by sequestering YAP/TAZ from TEAD, whereas other subunits including ACTL6A and BRM are thought to promote YAP/TAZ transcriptional activity by enhancing chromatin accessibility at YAP/TAZ/TEAD bound sites. Figure created with BioRender.com.
Tumor heterogeneity, functional degeneracy, and lineage plasticity are factors that reduce drug efficacy and lead to acquired resistance. Recent studies have established the role of Hippo signaling network at the epicenter of cancer-associated transcriptional addiction and drug resistance. Thus, mechanistic understanding of cross talks between YAP/TAZ/TEAD with other TFs and chromatin remodelers may reveal gene regulatory networks that drive tumor invasion and growth.
2. TEADs
TEAD TFs (TEAD1-4) are evolutionarily conserved from Drosophila to humans. They regulate developmental processes transcending a wide variety of tissue types, from the formation of neural tubes to the development of heart, brain, and skeletal muscle. Several chromatin immunoprecipitation (ChIP) studies reveal that TEADs serve as the primary effectors of the YAP/TAZ signaling: (1) 78% of the TEAD4-bound promoters and enhancers are co-occupied by YAP/TAZ in NF2-null breast cancer cell line MDA-MB-231 [12]; (2) YAP and TEAD1 co-occupy >80% of the promoters in MCF10A breast cancer cells [13]; and (3) 86% of all the YAP1 peaks contain at least one TEAD-binding site in SF268 glioblastoma cell line [14]. TEADs bind to the DNA but are barely known to exert any transcriptional activity by themselves [15]. They form complexes with multiple TFs, coactivators, and chromatin remodelers to regulate gene expression in diseases and cancers. Despite the crucial role they play in tumorigenesis, the underlying molecular mechanism of TEAD-mediated transcriptional regulation is not well understood. TEADs share >80% homology in the DNA-binding domain and >70% homology in the cofactor-binding domain amongst themselves [16]. Despite having a high degree of sequence similarity, they control different sets of tissue-specific enhancer elements. The role of YAP/TAZ/TEAD as regulators of gene expression through distal enhancers and in cofactor switching has attracted significant interest in recent years, but the differential regulation of the individual TEADs and its significance in this emerging paradigm of transcriptional addiction in cancer largely remains unknown. They are also not known to harbor any oncogenic mutations.3. YAP/TAZ/TEAD Control Gene Expression from Enhancers
Previously, it was thought that YAP/TAZ are coactivators for promoter driven transcription of a limited number of target genes [13]. However, recent technological advancement in the field of genome-wide ChIP seq and Transposase-Accessible Chromatin with high-throughput sequencing (ATAC-seq) have underscored the ability of YAP/TAZ/TEAD to tether to the chromatin at distal enhancer regions [12][14][17]. The importance of YAP/TAZ-mediated gene regulation through distant regulatory elements has shed light onto the global organization of relevant genes within the chromatin. TEADs are the primary mediators of YAP/TAZ recruitment to the chromatin. Interestingly, only 3.6% of the YAP/TAZ/TEAD binding is located in the promoter regions, in contrast to 91% of the complex located in the enhancer region, which can regulate expression of distal genes by chromatin looping [12]. ChIP data from various cell lines of different lineages and genetic backgrounds—such as glioblastoma line SF268 (YAP amplification), malignant mesothelioma line NCI-H2052 (NF2 mutation and LATS2 deletion), and non-transformed cells (IMR90)—has revealed that only a small set of YAP1/TEAD peaks are found within the gene transcription start sites (TSSs) while the majority are located at the distal active enhancers marked by histone post-translational modifications, including H3K27ac. The fact that YAP is required to maintain H3K27ac levels and TEAD occupancy at the YAP1-bound enhancer signifies its importance in establishing proper chromatin structures via positive feedback loop. Thus, YAP/TEAD distal binding is a general characteristic feature that tightly regulates transcription of a myriad of genes in both normal and cancer cells [14]. YAP further recruits mediator complexes to active enhancer sites, which can in turn regulate transcription via recruitment of Cyclin-Dependent Kinase 9 (CDK9), instilling “super enhancer”-like characteristics. Recruitment of CDK9 releases paused RNA Polymerase II (RNAPolII) at the promoters and induces elongation of YAP/TAZ targets to drive tumorigenesis in liver cancer cells. In the model liver cancer cell line HuCCT1, YAP occupies only 7% of the sites bound by TEADs, indicating that these super enhancer-like sites form a small subset of modules that are heavily overloaded with cofactors and activating histone marks. They play an important role in driving rapid gene expression in response to physiological and oncogenic cues and may play a role in YAP/TAZ-dependent cancer addiction as well [18]. In agreement with these studies, overexpression of the constitutively active form of YAP (YAP5SA) has been shown to reprogram cardiomyocytes into a fetal-like state by engaging a subset of global enhancers that are enriched for TEAD and AP-1 motifs. YAP overexpression promotes chromatin looping, which in turn increases enhancer–promoter contacts to access new target genes involved in developmental processes [19]. In flies, Yki can recruit mediator and histone methytranferase (HMT) complex via Ncoa6, a subunit of the Thritorax-related (Trr) methyltransferase complex to the promoter sites [20][21]. However, it is not known if Yki can engage in distal enhancer regulated gene expression. Ncoa6 can in turn regulate the expression of a subset of YAP targets in mammals, as reported in H69 cells. The regulation of enhancers by YAP/TAZ/TEAD is relevant to cancer as well. Recent studies reported enrichment of YAP/TAZ-bound enhancers in patient derived organoid models of human colorectal cancer. These enhanceosomes are conserved across different patient-derived tumor types with a range of genetic alterations, serving as a common node where several oncogenic signaling converge to drive tumorigenesis [17]. It still remains a major challenge to correlate target gene expression with a specific set of enhancers. New technological advances in single cells can address this issue (covered in later section) and answer questions in this emerging field of enhancer deregulation in YAP driven cancer and their role in therapeutic resistance.Table 1. Nuclear effectors of YAP/TAZ/TEAD that play a role in YAP-mediated transcriptional addictions and drug resistance in cancers.
| Factors | Conclusion | Tissue Origin | Reference |
|---|---|---|---|
| AP-1 and STAT | YAP/TAZ/TEAD and AP-1 transcription factors bind at the at the same genomic loci harboring TEAD and AP-1 composite sites. AP-1 enhances YAP/TAZ-induced oncogenic growth. | Breast | [12] |
| TEAD and AP-1 co-occupy the cis-regulatory region. TEAD/AP-1 engages with steroid receptor c-activators 1-3 (SRC1-3) to regulate migration and invasion. | Brain, colon, lung, endometrium | [22] | |
| Vemurafenib (small-molecule inhibitor of BRAF V600E)-induced drug resistance is partially mediated by the activity of JUN and/or AP-1 and TEAD. | Skin | [23] | |
| AP-1 drives YAP-dependent transformations. | Skin, pancreas | [24][25] | |
| AP-1 is a transcriptional target of YAP/TAZ; induced AP-1 can collaborate with YAP/TAZ to promote organ growth. | Liver | [26] | |
| FOSL1/AP-1 acts as a common node in MAPK and Hippo pathways. | Colon and lung pharynx, esophagus, cervix, ovary | [27][28] | |
| YAP/TAZ are recruited by different forms of TEAD/STAT3/AP-1 complex depending on the cis-recruiting motifs to regulate different sets of YAP/TAZ target genes. | Breast | [29] | |
| ERα/FOXA1 | YAP/TEAD act as ERα cofactors to regulate ERα-bound enhancer activation by recruiting MED1. | Breast | [30] |
| BRD4 | Enhancers occupied by YAP–TAZ show enrichment for BRD4, displaying super-enhancer-like characteristics and thus being sensitive to JQ1. | Breast | [31] |
| ARID1A sequesters YAP/TAZ from binding to TEAD to decrease YAP/TAZ activity. | Liver | [32] | |
| SWI/SNF | Pan-FGFR inhibition represses chromatin loading of BRG1, causing an epigenetic switch to promote YAP transcriptional dependency. | Breast | [33] |
| Increased ACTL6A promotes loading of TEAD-YAP binding to BAF complexes, which can enhance co-binding of each other to the chromatin through a positive feedback loop. | Pharynx, lung, esophagus (squamous cells) | [34] |
4. Role of AP-1 and STAT in YAP/TAZ/TEAD Transcriptional Regulation
Activator protein-1 (AP-1) is a well-characterized heterodimeric transcription factor, comprising two families of oncoproteins, namely, FOS and Jun. FOS family of proteins are early gene products and are rapidly induced in response to many cellular and extracellular cues, including cellular stress, developmental cues, and growth factors, as well as mitogens such as serum and lysophosphatidic acid (LPA) phorbol esters, which are also known to activate YAP activity [35]. The FOS family of proteins can regulate YAP activation and YAP/TAZ-derived phenotypes, such as cell survival, proliferation, and differentiation. Increasing evidence suggests that transcription control by AP-1 and YAP is highly interwoven and AP-1 and TEADs can synergistically drive tumor growth across several tumor types. In the NF2-null breast cancer cell line (MDA-MB-231), 70% of the YAP/TAZ/TEAD-occupied enhancers also contained AP-1-binding motifs, making AP-1 the second most abundant motif after TEAD. Sequential ChIP seq analysis for YAP followed by JUN suggested that both TEAD and AP-1 can bind to the cis-regulatory elements bearing TEAD and AP-1 composite sites at the same time and can physically interact with each other. AP-1 synergizes with YAP to increase oncogenic growth in mammary cells via activating target genes that control S-phase entry and mitosis. AP-1 has elevated activity in skin tumorigenic induced by chemical carcinogenesis. YAP/TAZ-deficient mice failed to produce tumors when subjected to chemical carcinogenesis, underscoring the importance of YAP/TAZ in AP-1-mediated tumorigenesis [12]. Since YAP/TAZ rapidly translocate to the nucleus upon serum or LPA stimulation, they act as immediate sensors regulating early gene expression. Recent studies have shown that AP-1 is a direct transcriptional target of YAP/TAZ and is induced in response to mitogenic signals. Knocking out YAP in HEK293 cells severely reduces the expression of AP-1 upon LPA treatment, whereas overexpressing a constitutively active form of YAP (5SA-YAP) induces AP-1 expression under serum starvation. In addition, the induction of FOS expression is dependent on TEAD binding to YAP, which can then synergize with YAP/TAZ to drive target gene expression. A mutant version of YAP with defective TEAD binding (S94AYAP) fails to rescue AP-1 expression in YAP/TAZ KO cells. Consistent with these observations, inhibition of AP-1 significantly rescued YAP-mediated liver overgrowth in mice, highlighting a previously unknown role of AP-1/YAP/TAZ in the regulation of immediate early gene expression and organ growth [26]. There has also been evidence of YAP/AP-1 collaboration in pancreatic cancer progression. For example, deletion of LATS1/2 in organoids or in a mouse model of pancreatic cancer led to the activation of AP-1 and YAP target genes, where YAP physically interacts with the FOS/JUN complex. Concomitantly, treatment with AP-1 inhibitors reduces YAP-mediated transformations [25]. Interestingly, YAP is dispensable for normal epidermal homeostasis in comparison to basal cell carcinoma (BCCs). Mice containing a conditional KO of YAP have normal epidermis; however, YAP/TEAD promotes BCC growth by inducing AP-1 signaling. A loss in the level of YAP severely affected AP-1 family transcription factor c-JUN in BCCs, decreasing its stability and transcriptional activity. YAP transcriptional regulation of AP-1 factors is also evolutionarily conserved in Drosophila. Activating transcription factor 3 (Atf3) is a direct transcriptional target of Sd, which is significantly upregulated in Ras driven tumor formation. Tumor specific gene expression in Drosophila is tightly regulated by a few key transcription factors, and AP-1 forms one of the major regulatory nodes. Loss of AP-1 or STATS can break this regulatory network by reducing the expression of tumor signature genes [36]. In the mammalian system, transcription of AP-1 is also affected by YAP as both TEAD and AP-1 bind to the promoter and enhancer sites, forming an autoregulatory loop. YAP impacts the transcription of known components of MAPK signaling in BCCs, but the upstream molecular mechanisms that connect YAP with the JNK-JUN axis are still not well understood [24]. Furthermore, it has been shown that TEAD and AP-1 co-occupy cis-regulatory region across a broad range of tumor cells such as the colon, lung, neuroblastoma, and endometrial cancer. TEAD/AP-1 transcription factors can engage with steroid receptor c-activators 1-3 (SRC1-3), which bridge between TEAD and AP-1 to regulate migration and invasion. SRC inhibition significantly inhibits the interaction of JUND with TEAD [22]. A recent study from one lab focused on an integrated strategy combining machine learning with chemicogenomics, to identify lineage-independent gene signatures for Hippo pathway in pan-cancer cell lines. It can be observed crosstalk between Hippo and MAPK signaling, converging at the level of AP-1 and serving as a common node to regulate gene expression. FOSL1 can interact with TEADs in the presence of YAP1. ATAC-seq under conditions of YAP depletion combined with MEK inhibition revealed decreased chromatin accessibility at TEAD and AP-1 binding sites [28]. Several studies show that YAP/TAZ activation serves as a bypass mechanism to overcome RAS/MAPK blockade. YAP1 rescues KRAS suppression in KRAS-dependent cancer cells. Tumors that escape KRAS suppression in a KRAS-driven murine lung cancer model show high YAP1 activity and upregulation of epithelial-to-mesenchymal transition (EMT)-like transcriptional programs. These gene signatures are jointly regulated by YAP1 and FOS, where YAP1 physically interacts with FOS at the promoters to drive YAP-induced transformation. YAP/TAZ activation also plays a crucial role in acquiring drug-induced resistance in BRAF and KRAS mutant cancer cells treated with EGFR/MAPK inhibitors. JUN and/or AP-1 and TEAD has been shown to induce resistance to vemurafenib (small-molecule inhibitor of BRAF V600E) in cultured patient-derived melanoma cells [23]. However, it still remains to be investigated whether AP-1-mediated cell cycle and EMT transcriptional programs might be one of the mechanisms to promote resistance to these other targeted therapies [27]. YAP/TAZ also act as a transcriptional coactivator for JUNB and STAT, promoting cellular transformation. YAP/TAZ, but not JUNB, are required for STAT3 phosphorylation during breast epithelial cell transformation. Along with TEAD/AP-1, YAP/TAZ target genes are also associated with STAT3, albeit to a lesser extent. YAP/TAZ are recruited by different forms of TEAD/STAT3/AP-1 complex depending on the cis-recruiting motifs, which can then regulate different sets of YAP/TAZ target genes. Interaction with these transcription factors is cell-type-dependent, causing specific gene expression profiles with distinct functions [29]. There is limited evidence on whether expression of YAP is regulated by other AP-1-like TFs. A recent study showed that YAP1 is a downstream effector of MAPK signaling activated by the FGFR axis, which promotes gastric cancer (GC) progression. JUN physically interacts with YAP1/TEAD4 complex and, consequently, knockdown of JUN impairs YAP1/TEAD4 complex formation, leading to reduced cell proliferation in model cell lines. FGFR activation promotes cJUN phosphorylation, and knockdown of FGFR significantly decreases YAP mRNA expression; however, the exact molecular mechanism is still unclear [37]. Further work is necessary to tease apart the synergistic roles from specialized roles of AP-1 and TEADs, as well as to assess the potential of AP-1 to be a therapeutic target for YAP/TAZ-dependent cancer. Moreover, it would be interesting to know whether this de novo induction of AP-1 expression is required for all or some of YAP/TAZ biological functions.References
- Johnson, R.; Halder, G. The Two Faces of Hippo: Targeting the Hippo Pathway for Regenerative Medicine and Cancer Treatment. Nat. Rev. Drug Discov. 2014, 13, 63–79.
- Manning, S.A.; Kroeger, B.; Harvey, K.F. The Regulation of Yorkie, YAP and TAZ: New Insights into the Hippo Pathway. Development 2020, 147, dev179069.
- Oh, H.; Irvine, K.D. In Vivo Regulation of Yorkie Phosphorylation and Localization. Development 2008, 135, 1081–1088.
- Luo, J.; Yu, F.-X. GPCR-Hippo Signaling in Cancer. Cells 2019, 8, 426.
- Cai, X.; Wang, K.-C.; Meng, Z. Mechanoregulation of YAP and TAZ in Cellular Homeostasis and Disease Progression. Front. Cell Dev. Biol. 2021, 9, 673599.
- Plouffe, S.W.; Meng, Z.; Lin, K.C.; Lin, B.; Hong, A.W.; Chun, J.V.; Guan, K.-L. Characterization of Hippo Pathway Components by Gene Inactivation. Mol. Cell 2016, 64, 993–1008.
- Zheng, Y.; Wang, W.; Liu, B.; Deng, H.; Uster, E.; Pan, D. Identification of Happyhour/MAP4K as Alternative Hpo/MST-like Kinases in the Hippo Kinase Cascade. Dev. Cell 2015, 34, 642–655.
- Kim, M.-K.; Jang, J.-W.; Bae, S.-C. DNA Binding Partners of YAP/TAZ. BMB Rep. 2018, 51, 126–133.
- Totaro, A.; Panciera, T.; Piccolo, S. YAP/TAZ Upstream Signals and Downstream Responses. Nat. Cell Biol. 2018, 20, 888–899.
- Calses, P.C.; Crawford, J.J.; Lill, J.R.; Dey, A. Hippo Pathway in Cancer: Aberrant Regulation and Therapeutic Opportunities. Trends Cancer Res. 2019, 5, 297–307.
- Nguyen, C.D.K.; Yi, C. YAP/TAZ Signaling and Resistance to Cancer Therapy. Trends Cancer Res. 2019, 5, 283–296.
- Zanconato, F.; Forcato, M.; Battilana, G.; Azzolin, L.; Quaranta, E.; Bodega, B.; Rosato, A.; Bicciato, S.; Cordenonsi, M.; Piccolo, S. Genome-Wide Association between YAP/TAZ/TEAD and AP-1 at Enhancers Drives Oncogenic Growth. Nat. Cell Biol. 2015, 17, 1218–1227.
- Zhao, B.; Ye, X.; Yu, J.; Li, L.; Li, W.; Li, S.; Yu, J.; Lin, J.D.; Wang, C.-Y.; Chinnaiyan, A.M.; et al. TEAD Mediates YAP-Dependent Gene Induction and Growth Control. Genes Dev. 2008, 22, 1962–1971.
- Stein, C.; Bardet, A.F.; Roma, G.; Bergling, S.; Clay, I.; Ruchti, A.; Agarinis, C.; Schmelzle, T.; Bouwmeester, T.; Schübeler, D.; et al. YAP1 Exerts Its Transcriptional Control via TEAD-Mediated Activation of Enhancers. PLoS Genet. 2015, 11, e1005465.
- Xiao, J.H.; Davidson, I.; Matthes, H.; Garnier, J.M.; Chambon, P. Cloning, Expression, and Transcriptional Properties of the Human Enhancer Factor TEF-1. Cell 1991, 65, 551–568.
- Holden, J.K.; Cunningham, C.N. Targeting the Hippo Pathway and Cancer through the TEAD Family of Transcription Factors. Cancers 2018, 10, 81.
- Della Chiara, G.; Gervasoni, F.; Fakiola, M.; Godano, C.; D’Oria, C.; Azzolin, L.; Bonnal, R.J.P.; Moreni, G.; Drufuca, L.; Rossetti, G.; et al. Epigenomic Landscape of Human Colorectal Cancer Unveils an Aberrant Core of Pan-Cancer Enhancers Orchestrated by YAP/TAZ. Nat. Commun. 2021, 12, 2340.
- Galli, G.G.; Carrara, M.; Yuan, W.-C.; Valdes-Quezada, C.; Gurung, B.; Pepe-Mooney, B.; Zhang, T.; Geeven, G.; Gray, N.S.; de Laat, W.; et al. YAP Drives Growth by Controlling Transcriptional Pause Release from Dynamic Enhancers. Mol. Cell 2015, 60, 328–337.
- Monroe, T.O.; Hill, M.C.; Morikawa, Y.; Leach, J.P.; Heallen, T.; Cao, S.; Krijger, P.H.L.; de Laat, W.; Wehrens, X.H.T.; Rodney, G.G.; et al. YAP Partially Reprograms Chromatin Accessibility to Directly Induce Adult Cardiogenesis In Vivo. Dev. Cell 2019, 48, 765–779.e7.
- Oh, H.; Slattery, M.; Ma, L.; Crofts, A.; White, K.P.; Mann, R.S.; Irvine, K.D. Genome-Wide Association of Yorkie with Chromatin and Chromatin-Remodeling Complexes. Cell Rep. 2013, 3, 309–318.
- Oh, H.; Slattery, M.; Ma, L.; White, K.P.; Mann, R.S.; Irvine, K.D. Yorkie Promotes Transcription by Recruiting a Histone Methyltransferase Complex. Cell Rep. 2014, 8, 449–459.
- Liu, X.; Li, H.; Rajurkar, M.; Li, Q.; Cotton, J.L.; Ou, J.; Zhu, L.J.; Goel, H.L.; Mercurio, A.M.; Park, J.-S.; et al. Tead and AP1 Coordinate Transcription and Motility. Cell Rep. 2016, 14, 1169–1180.
- Shaffer, S.M.; Dunagin, M.C.; Torborg, S.R.; Torre, E.A.; Emert, B.; Krepler, C.; Beqiri, M.; Sproesser, K.; Brafford, P.A.; Xiao, M.; et al. Rare Cell Variability and Drug-Induced Reprogramming as a Mode of Cancer Drug Resistance. Nature 2017, 546, 431–435.
- Maglic, D.; Schlegelmilch, K.; Dost, A.F.; Panero, R.; Dill, M.T.; Calogero, R.A.; Camargo, F.D. YAP-TEAD Signaling Promotes Basal Cell Carcinoma Development via a c-JUN/AP1 Axis. EMBO J. 2018, 37, e98642.
- Park, J.; Eisenbarth, D.; Choi, W.; Kim, H.; Choi, C.; Lee, D.; Lim, D.-S. YAP and AP-1 Cooperate to Initiate Pancreatic Cancer Development from Ductal Cells in MiceYAP and AP-1 Initiate PDAC from Ductal Cells. Cancer Res. 2020, 80, 4768–4779.
- Koo, J.H.; Plouffe, S.W.; Meng, Z.; Lee, D.-H.; Yang, D.; Lim, D.-S.; Wang, C.-Y.; Guan, K.-L. Induction of AP-1 by YAP/TAZ Contributes to Cell Proliferation and Organ Growth. Genes Dev. 2020, 34, 72–86.
- Shao, D.D.; Xue, W.; Krall, E.B.; Bhutkar, A.; Piccioni, F.; Wang, X.; Schinzel, A.C.; Sood, S.; Rosenbluh, J.; Kim, J.W.; et al. KRAS and YAP1 Converge to Regulate EMT and Tumor Survival. Cell 2014, 158, 171–184.
- Pham, T.H.; Hagenbeek, T.J.; Lee, H.-J.; Li, J.; Rose, C.M.; Lin, E.; Yu, M.; Martin, S.E.; Piskol, R.; Lacap, J.A.; et al. Machine-Learning and Chemicogenomics Approach Defines and Predicts Cross-Talk of Hippo and MAPK PathwaysMachine-Learning Approach Predicts Hippo Pathway Dependency. Cancer Discov. 2021, 11, 778–793.
- He, L.; Pratt, H.; Gao, M.; Wei, F.; Weng, Z.; Struhl, K. YAP and TAZ Are Transcriptional Co-Activators of AP-1 Proteins and STAT3 during Breast Cellular Transformation. Elife 2021, 10, e67312.
- Zhu, C.; Li, L.; Zhang, Z.; Bi, M.; Wang, H.; Su, W.; Hernandez, K.; Liu, P.; Chen, J.; Chen, M.; et al. A Non-Canonical Role of YAP/TEAD Is Required for Activation of Estrogen-Regulated Enhancers in Breast Cancer. Mol. Cell 2019, 75, 791–806.e8.
- Zanconato, F.; Battilana, G.; Forcato, M.; Filippi, L.; Azzolin, L.; Manfrin, A.; Quaranta, E.; Di Biagio, D.; Sigismondo, G.; Guzzardo, V.; et al. Transcriptional Addiction in Cancer Cells Is Mediated by YAP/TAZ through BRD4. Nat. Med. 2018, 24, 1599–1610.
- Chang, L.; Azzolin, L.; Di Biagio, D.; Zanconato, F.; Battilana, G.; Lucon Xiccato, R.; Aragona, M.; Giulitti, S.; Panciera, T.; Gandin, A.; et al. The SWI/SNF Complex Is a Mechanoregulated Inhibitor of YAP and TAZ. Nature 2018, 563, 265–269.
- Li, Y.; Qiu, X.; Wang, X.; Liu, H.; Geck, R.C.; Tewari, A.K.; Xiao, T.; Font-Tello, A.; Lim, K.; Jones, K.L.; et al. FGFR-Inhibitor-Mediated Dismissal of SWI/SNF Complexes from YAP-Dependent Enhancers Induces Adaptive Therapeutic Resistance. Nat. Cell Biol. 2021, 23, 1187–1198.
- Chang, C.-Y.; Shipony, Z.; Lin, S.G.; Kuo, A.; Xiong, X.; Loh, K.M.; Greenleaf, W.J.; Crabtree, G.R. Increased ACTL6A Occupancy within MSWI/SNF Chromatin Remodelers Drives Human Squamous Cell Carcinoma. Mol. Cell 2021, 81, 4964–4978.e8.
- Yu, F.-X.; Zhao, B.; Panupinthu, N.; Jewell, J.L.; Lian, I.; Wang, L.H.; Zhao, J.; Yuan, H.; Tumaneng, K.; Li, H.; et al. Regulation of the Hippo-YAP Pathway by G-Protein-Coupled Receptor Signaling. Cell 2012, 150, 780–791.
- Atkins, M.; Potier, D.; Romanelli, L.; Jacobs, J.; Mach, J.; Hamaratoglu, F.; Aerts, S.; Halder, G. An Ectopic Network of Transcription Factors Regulated by Hippo Signaling Drives Growth and Invasion of a Malignant Tumor Model. Curr. Biol. 2016, 26, 2101–2113.
- Zhang, J.; Wong, C.C.; Leung, K.T.; Wu, F.; Zhou, Y.; Tong, J.H.M.; Chan, R.C.K.; Li, H.; Wang, Y.; Yan, H.; et al. FGF18-FGFR2 Signaling Triggers the Activation of c-Jun-YAP1 Axis to Promote Carcinogenesis in a Subgroup of Gastric Cancer Patients and Indicates Translational Potential. Oncogene 2020, 39, 6647–6663.
More