Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Catherine Yang and Version 1 by Feng Ru Tang.
Radiation-induced brain injury (RIBI) after radiotherapy has become an increasingly important factor affecting the prognosis of patients with head and neck tumor. With the delivery of high doses of radiation to brain tissue, microglia rapidly transit to a pro-inflammatory phenotype, upregulate phagocytic machinery, and reduce the release of neurotrophic factors.
- microglia
- brain injury
- ionizing radiation
- cognitive effects
- therapy
1. Introduction
Radiotherapy is the mainstay of first-line treatment in primary and metastatic brain tumors [1]. Unfortunately, irradiated areas always include normal brain tissue that surrounds the tumor, and as a result, many patients experience progressive and irreversible side effects. At the early stage after radiotherapy, patients may have transient, self-healing symptoms, including headache, lethargy, fatigue, and attention deficits, whereas more than 50% of oncology patients who survive more than 6 months after whole-brain radiation develop irreversible cognitive impairment [2,3,4,5][2][3][4][5]. The molecular and cellular mechanisms behind these effects are complex, involving the production of proinflammatory factors from microglia, cascades of signal transduction, gliosis, altered neurogenesis, and injury of endothelial cells (ECs) [6,7][6][7]. Currently, multiple radiation strategies that limit normal tissue toxicity such as hippocampal avoidance radiotherapy, proton beam therapy, and ultra-high-dose-rate irradiation, have been shown to moderate RIBI in clinical or preclinical studies [8,9,10,11][8][9][10][11]. However, the lack of understanding of cellular responses to ionizing radiation (IR) in the central nervous system (CNS) has limited the development of new therapeutic approaches.
2. Microglia in Radiation-Induced Brain Injury
Following IR, microglia sense microenvironmental changes immediately and react accordingly. By interacting with neurons, ECs, astrocytes, and oligodendrocytes, these cells mediate brain pathogenesis, including BBB disruption, infiltration of peripheral immune cells, neuronal death, inhibition of neurogenesis, and structural damage of synapses [62,63,64,65][12][13][14][15].
2.1. Microglial Activation
DNA damage caused by IR is an important activator of microglia. High LET directly ionizes DNA molecules, while low LET tends to indirectly damage DNA through ROS and free radicals originating from water radiolysis [6,66,67][6][16][17]. Damaged DNA can quickly trigger the activation of transcription factors such as nuclear factor κB (NF-κB), cAMP response element-binding protein (CREB), and activating protein 1 (AP-1), which control intracellular ROS generation and gene expression of inflammatory factors including IL-1β, TNF-α, cyclooxygenase 2 (COX-2), and monocyte chemoattractant protein-1 (MCP-1/CCL2) [18,68][18][19]. While healthy neurons release factors that inhibit microglial activation, radiation-induced damaged or dead neurons reduce this inhibition and increase the production of various chemokines, cytokines, reactive oxygen species, and ATP [13,63,69][13][20][21]. By virtue of abundant receptors on their cell membrane, microglia sense and respond to the changes of “danger” signals in the surrounding environment. For instance, high mobility group box 1 (HMGB1) from neurons or ECs and microglial toll-like receptor 4 (TLR4) expression were upregulated after IR, and the combination of both promoted microglial activation [70,71][22][23]. Peripheral immune cells infiltrate the brain tissue following radiation-induced damage to the BBB, and they produce ROS, which in turn activates microglia [3].
Once activated, microglia move towards the injury site, phagocytose apoptotic neurons and cell debris, and produce large amounts of pro-inflammatory mediators [72][24]. It was shown that following in vivo or in vitro irradiation higher than 7 Gy, microglia produced high levels of ROS, NO, IL-1, TNF-α, IL-6, COX-2, MCP-1, and intercellular adhesion molecule 1 (ICAM-1) [20,62,73,74,75,76][12][25][26][27][28][29]. These pro-inflammatory mediators exacerbated RIBI. Studies in rodents have also shown that activated microglia and TNF-α remained at high levels for at least 6 months after a single high dose of irradiation [77,78][30][31]. Such persistently activated microglia continuously release pro-inflammatory factors, which maintain the inflammatory status of the brain microenvironment and further inflict neuronal and progenitor cell death, leading to a vicious circle characterized by microglial activation, release of inflammatory factors, and neuronal death [79][32]. Persistent inflammation also inhibits neurogenesis in the juvenile and adult hippocampus as X-ray irradiation with 2 Gy at postnatal day 10 induces impairment of neurogenesis, even when animals are six month old [80][33]. X-ray irradiation of adult mice with 10 Gy suppressed the proliferative capacity of neural progenitor cells (NPCs) in the DG region and induced NPCs to differentiate towards glial cells, which was attributed to an inflammatory response, as aggressive anti-inflammatory strategies partially restored the proliferative capacity of NPC to neurons. Importantly, cognitive and behavioral modifications correspond with increased microglial activation, and administration of anti-inflammatory agents also reduced cognitive impairment in rodent [65,79,80][15][32][33]. It should be noted that factors affecting microglia activation, such as age, gender, environment, and cell interactions with microglial cells, and expression and activation of different receptor on microglia may affect the efficacy of anti-inflammatory strategies [19][34].
2.2. ROS/RNS Production and Oxidative Stress
A delicate balance between reactive oxygen/nitrogen and antioxidants is essential for the maintenance of normal physiological function of the CNS. In RIBI, the disruption of equilibrium often means the excessive accumulation of ROS/RNS in the cell, leading to lipid peroxidation, protein degradation, and DNA damage reactions [81][35]. Microglia respond to the pathogen- and stressors-associated molecular patterns through the production of ROS and protect normal tissues from insults [81,82][35][36]. However, high doses of IR induce excess ROS production, which is further amplified with increasing radiation doses [83,84][37][38].
Enzymatic and non-enzymatic reactions are the main pathways of ROS production. After radiation exposure, non-enzymatic ROS are generated in large amounts along with mitochondrial respiration of microglia [84][38]. NADPH oxidase (NOX) consists of Nox1 to 5 and dual oxidases 1 and 2 and promotes enzymatic ROS production in most cells [85][39]. Microglia express high levels of NOX, particularly NOX-2 [86][40]. NOX-2 expression is significantly elevated in the brain within hours after IR, and the NOX-2 inhibitors apocynin and diphenylene iodonium, or the neutralizing antibody to NOX-2 significantly reduced radiation-induced ROS production [87][41]. In irradiated microglia, NOX activation-mediated ROS production is modulated by the mitogen-activated protein kinases (MAPKs) signaling cascade through phosphorylation of c-Jun, a component of AP-1 transcription factors [88,89][42][43]. Mitochondrial translocator protein 18 kDa (TSPO) is located on the outer mitochondrial membrane, similar to NOX2, and it is upregulated in reactive microglia [90,91][44][45]. It has been shown that gamma irradiation with 2 Gy upregulated TSPO expression in primary microglia [90][44]. TSPO is associated with ROS generation and subsequent oxidative stress. Stimulation of primary microglia with TSPO typical ligands PK11195 and Ro5-4864 induced ROS production, and prior application of a NOX inhibitor reversed this effect [92][46]. Two recent studies have revealed more details on this interaction between TSPO and NOX in microglial ROS production [93,94][47][48]. In mice with selective deletion of TSPO in microglia, it has been demonstrated that TSPO-mediated ROS generation is Nox1 dependent in reactive microglia, and an increase in cytosolic calcium concentrations is necessary for functional coupling between TSPO and NOX-1 [93][47]. On the other hand, TSPO interacts with NOX2 subunits gp91Phox and p22Phox in resting microglia. This interaction is disrupted after endotoxin exposure, resulting in upregulation of TSPO at the mitochondria and plasma membrane, which provided a biophysical foundation for their interaction that regulates ROS production under radiation conditions [94][48]. In addition to affecting mitochondria-associated oxidative stress, TSPO also affects the microglial genomic function. TSPO is involved in inflammatory transcriptional programs, including NLRP3 inflammasome, NF-κB, and MAPK [95[49][50][51],96,97], leading to the release of multiple cytokines. In primary human, mouse, and rat microglia, PK11195 has been shown to inhibit LPS-induced production of inflammatory factors such as TNF-α, IL-6, and NO [98,99,100,101][52][53][54][55]. Recently, treatment of microglia with the new generation TSPO ligands 2-cl-mgv-1 and mgv-1 also reduced the production of COX2, iNOS, and NO after LPS stimulation [101,102][55][56].
Another cause of ROS accumulation is dysregulation of complex antioxidant systems. Microglia contain superoxide dismutase, catalase, and NADPH-regenerating enzymes, as well as a high concentration of glutathione and enzymes necessary to generate glutathione, which confer high antioxidant activity to these cells. A single dose of more than 2 Gy significantly reduces the activity of antioxidant enzymes, including superoxide dismutase (SOD), glutathione, and catalase [103,104,105][57][58][59].
ROS derived from NOX and mitochondria may underlie radiation-induced excessive inflammation. There is ample evidence that enzymatic ROS promotes the release of proinflammatory factors in microglia, and both direct inhibition of NOX-2 and elimination of NOX-2-dependent ROS production reduce the expression of pro-inflammatory factors in microglia [106,107][60][61]. Indeed, as the first messenger of intercellular communication, ROS released by microglia change the redox state of adjacent cells [108,109][62][63]. On the other hand, increased ROS, as second messengers, through affecting the activation of kinase pathways and transcription factors, promote microglial immune activation, and subsequently amplify and perpetuate neuroinflammation [110][64]. For instance, ROS could directly react with IκB kinase, which inhibits NF-κB activity or catalyzes the release of NF-κB subunit from the IκB binding state through redox activation of upstream kinases, thereby initiating the expression of pro-inflammatory genes in microglia [111,112][65][66]. Co-incubation of BV-2 cells with PPARδ agonist suppressed the radiation-induced increase in intracellular ROS generation to reduce NF-κB and AP-1 activation and inflammatory factor gene expression [18]. Limiting mitochondrial ROS accumulation with mitoTEMPO suppressed MAPKs activation and nuclear translocation of NF-κB, accompanied by the reduced expression of different proinflammatory factors, such as TNF-α, IL-1β, IL-6, iNOS, and COX-2 [113][67]. The peak of radiation-induced ROS production precedes IL-1, TNF-α, COX-2, and MCP-1 in microglia [18,20][18][25]. After fractionated whole-brain irradiation, the production of ROS peaks at 4 h after radiation, whereas protein levels of TNF-α and MCP-1 are significantly increased at 8 h after radiation [87][41]. Therefore, radiation-induced ROS may be an important cause for the subsequent occurrence of pro-inflammatory events in RIBI.
2.3. Regulation of BBB Integrity
BBB is composed of ECs, basal lamina, and astroglial end-feet. With highly selective permeability, BBB selects and controls the entry of most molecules from the circulating blood into the CNS [6]. In physiological conditions, perivascular microglia physically contact with ECs and monitor the passage of blood solutes through the BBB. Microglia also express tight junction protein claudin-5 to maintain tight-junction integrity between ECs [114,115][68][69]. So far, only a few studies have investigated the interaction of microglia with the BBB after IR exposure.
Following a single whole-brain irradiation with 20–60 Gy, an acute increase in BBB permeability was detected with the application of BBB permeable tracers. This BBB collapse is reversible and can be restored within weeks [64,116][14][70]. Although radiation at a dose of 10 Gy does not significantly damage BBB in mice, minor BBB permeability alterations may occur [117,118][71][72]. Fractioned-irradiation with a total dose of 40 Gy (2 Gy per fraction) for four weeks results in an increase in BBB permeability, which may last for 180 days [119][73]. Moreover, as a major trigger of increased BBB permeability, radiation-induced EC apoptosis increases with time and dose [3]. Irradiated ECs secrete cellular signals through the NF-κB pathway to activate microglia and attract microglia migration toward adjacent blood vessels [120,121][74][75]. Irradiation of isolated and co-culture systems show that astrocyte activation requires microglia-derived factors, including prostaglandin E2 (PGE2) [62][12]. An in vivo experiment also confirmed that radiation-induced astrocyte activation is medicated by C1q, which is produced by microglia [71][23]. In such a way, microglia and astrocytes exert synergistic effects to co-release proinflammatory cytokines, such as TNF-α and IL-6, which stimulate surviving ECs to upregulate their intercellular adhesion molecule 1 on the luminal surface of blood vessels [7,64,122][7][14][76]. Irradiated microglia can produce ICAM-1 directly or release TNF-α and IL-6 to activate astrocytes to produce ICAM-1 [123][77]. In response, peripheral leukocytes are recruited onto ECs and, along with microglia, secrete matrix metalloproteinases (MMPs) that break down the BBB, which then allows peripheral immune cells to enter the brain parenchyma and exacerbate brain damage [7,124][7][78]. In addition, activated microglia could downregulate claudin-5 expression via TNF-α production, which contributed to the radiation-induced early BBB disruption [64][14]. Anti-TNF-α treatment reduced BBB permeability and ICAM-1-dependent leukocyte adhesion in mice exposed to X-ray radiation with 20 Gy [125][79].
2.4. Immune Cell Infiltration in the Brain
Although microglia are innate immune cells in the brain, peripheral immune cells migrate into the brain due to disruption of BBB after high doses of ionizing radiation [126][80]. CD3+ cells infiltrate to brain tissues within 7 days after irradiation and stay there for 12 months, whereas penetration by CD11c+ and MHC II+ cells occurs at the late stage after 7 days. However, distinguishing peripherally infiltrated immune cells from resident microglia is difficult, as these two groups of cells express many identical immune markers, such as CD11c, CD 68, and MHC II [127][81]. With the identification of characteristic markers for microglia, the application of transgenic and bone marrow chimeric animals and experimental techniques, such as flow cytometry and two-photon imaging, identification, and functional investigation of infiltrating immune cells became feasible. Using bone marrow chimeric mice, the dose-dependent recruitment of bone marrow-derived (BMD) cells and their differentiation into inflammatory cells and microglia were demonstrated in the irradiated brain region [128][82]. This recruitment can persist up to 6 months after irradiation with doses above 15 Gy [129][83]. Mildner et al. identified a specific monocyte population that penetrated the brain and presented a microglia phenotype after cranial radiation [117][71]. Even in the absence of radiation-induced detectable BBB damage, blood-derived macrophages are recruited to the brain and express CX3CR1, a marker unique to microglia [117,118][71][72]. This recruitment without BBB damage may be a consequence of increased levels of adhesion molecules, chemokines, and their receptors associated with immune cell infiltration in the postradiation brain [118,130][72][84]. Among them, CCL2-CCR2 signaling has been shown to participate in this process. Irradiated microglia can secrete CCL2, but barely express CCR2 [131,132][85][86]. Several studies have reported that high doses of IR (≥9 Gy) caused increased levels of CCR2 + macrophages and CCL2 in the mouse brain parenchyma [71,118,133][23][72][87]. CCR2 deficiency reduced colonization of BMD immune cells into the brain 6 months after cranial radiation [130][84]. Interestingly, under relatively low-dose irradiation (doses below 2 Gy), CCR2 knockout mice exhibited preservation of survival NPCs in the hippocampus and improvement of spatial memory and learning deficits [134][88]. After a high dose of radiation (10 Gy), this protection afforded by CCR2 deficiency against cellular and behavioral deterioration was also identified [130][84]. These studies suggest that infiltrating cells may potentially exacerbate RIBI, although work remains to distinguish resident microglia and infiltrating immune cells. The expression of CCR2 in NPCs, granule cells, and pyramidal neurons after irradiation may also shift the researcher’s attention to the role of infiltrating cells in RIBI [135][89]. Dietrich et al. demonstrated that BMD macrophages and monocytes were chronically increased in the irradiated site and communicated with the cellular microenvironment where they existed perpetually, which reduced the inhibition of radiation on neuro-glial progenitor cell proliferation and improved cognitive function [136][90]. In summary, there are only a few RIBI models that investigate the functional roles of infiltrating cells. Since treatment that pharmacologically targets CNS microglia to prevent RIBI may also affect the survival, proliferation, and functional transitions of peripheral immune cells, further examination of the contributions of infiltrating cells to RIBI and their recruitment mechanisms is highly warranted.
3. Modulation of Microglia for RIBI Therapy
Many different biochemical mediators, their receptors, and downstream signaling pathways are involved in microglial reaction to RIBI (Figure 21). Inhibition or activation of these pathways may prevent RIBI (Table 1).
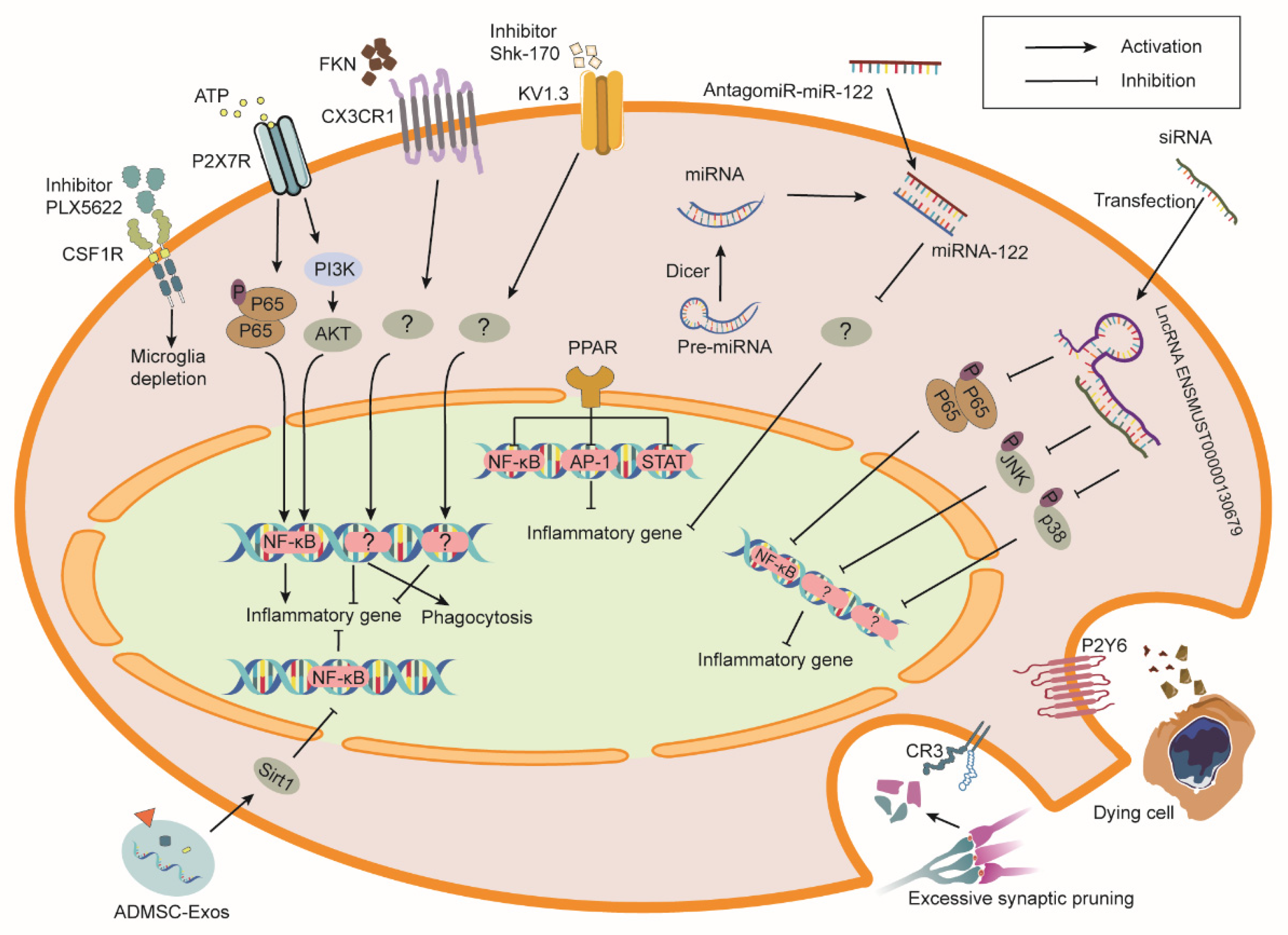
Figure 21. Schematic overview of receptors and exogenous molecules that modulate microglia phenotypes and functions in RIBI. Colony stimulating factor 1 receptor (CSF1R) blockade causes microglial death. Inhibition of P2X7 receptor (P2X7R) and KV1.3 channel or activation of (C-X3-C motif chemokine receptor 1) CX3CR1 and peroxisome proliferator-activated receptor (PPAR) prevent inflammatory gene expression in microglia. Activation of P2Y6 receptor (P2Y6R) and C3 receptor (CR3) mediate the phagocytosis of dying cells and synaptic components by microglia, respectively. Moreover, the introduction of exogenous molecules such as microRNA (miRNA), long non-coding RNAs (LncRNA), and extracellular vesicles (EVs) also enhance the therapeutic efficacy of RIBI. ATP, adenosine triphosphate; P, phosphorylation; NF-κB, nuclear factor κB; PI3K, phosphoinositide 3 kinases; AKT, protein kinase B; FKN, fractalkine; Shk-170, Stichodactyla helianthus-170; Pre-miRNA, precursor-miRNAs; siRNA, short interfering RNA; AP-1, activator protein 1; STAT, signal transducer and activator of transcription; Sirt1, sirtuin 1; ADMSC-Exos, exosomes derived from adipose mesenchymal stem cells.
Table 1.
Radioprotective effect of targeting different molecules in microglia in radiation-induced brain injury models.
| Targets | Animal/Cell Model | Source Dose and Dose Rate | Irradiated Site | Time Point after Radiation | Intervention Effect in Irradiation Models | Reference |
|---|---|---|---|---|---|---|
| CSFR1 | C57BL/6J mouse | X-ray with 9 cGy (1.10 Gy/min) |
whole brain | 3 days, 2 weeks, 6 weeks | CSFR1 inhibition reduces the increase in mRNA of inflammation markers (TLR9, SYK, CCL6, CD14, CLECL5a, TSLP, CCL5) and the number of activated microglia in hippocampus and ameliorates cognitive dysfunction. | [141][91] |
| 4He particles with 30 cGy (15–25 cGy/min) |
4–6 weeks | CSFR1 inhibition ameliorates cognitive dysfunction, reduces activated microglia population, and attenuates the increase in PSD-95 puncta but does not affect morphologic and electrophysiologic features of neurons. | [143][92] | |||
| 4He particles with 15 cGy (16.37 cGy/min) 50 cGy (16.95 cGy/min) 100 cGy (18.07 cGy/min) |
18–21 days and 90–100 days | CSFR1 inhibition improves long-term cognitive impairment and inflammatory response, decreases C5aR and LAMP-1, and increases synapsin-1. | [144][93] | |||
| γ ray with three fractions of 3.3 Gy | 1, 3 months | CSFR1 blockade reduces the numbers of activated microglia, suppresses monocyte accumulation in brain, and ameliorates cognitive dysfunction. | [142][94] | |||
| C1q | C57BL/6 mouse | γ-ray with 9 Gy (1.2 Gy/min) |
whole brain | 2, 24, 48 h; 1, 2, 3, 4 weeks | Deletion of C1q in microglia protects synaptic loss and reduces activation of microglia and astrocytes, as well as protein levels of TNF-a, IL-1ß, IL-6, IL-1α, CCL2, IL-18, and TLR4. | [71][23] |
| C3 | C57BL/6 mouse | X-ray with 8 Gy (2.3 Gy/min) |
whole brain | 6 h; 7 days; 2, 3, 4 weeks | C3 knockout improves task performance and increases activated microglia and proliferating cells in the granule cell layer. | [165][95] |
| C3R | C57BL/6J mouse | γ-ray with 10 Gy (1.17 Gy/min) |
whole brain | 30 days | CR3 blockade ameliorates behavior deficits in novel object recognition and the Lashley III maze, prevents dendritic spine loss, and increases CD11-positive microglia in hippocampus. | [166][96] |
| 30, 45 days | CR3 knockout prevents dendritic spine loss and increases activated microglia in hippocampus. | [145][97] | ||||
| P2Y6 | Balb/c mouse | β-ray with 30 Gy (3 Gy/min) |
whole brain | 1, 14, 30 days | P2Y6 receptor antagonism suppresses phagocytosis of irradiated microglia and increases the number of apoptotic neurons. | [182][98] |
| Primary microglia | β-ray with 8 Gy | 4, 12, 48 h | P2Y6 receptor antagonism suppresses phagocytosis of irradiated microglia and has no effect on the production of inflammatory mediators (TNF-α, IL-1β, IL-6, iNOS). | [182][98] | ||
| P2X7 | Balb/c mouse | β-ray with 30 Gy (3 Gy/min) |
whole brain | 3, 7, 14 days; 8 weeks | P2X7R blockade reduces the activated microglia population and neuron loss in the cortex. | [69][21] |
| Primary microglia | β-ray with 10 Gy (6 MeV/min) |
24, 48 h | P2X7R blockade reduces the activated microglia population and mRNA expression levels of IL-6, TNF-α, and COX-2. | [69][21] | ||
| CX3CR1 | C57BL/6J mouse | γ-ray with 10 Gy (2 Gy/min) |
whole brain | 3, 6, 12, 24, 48, 72 h; 1, 2, 4 weeks | FKN overexpression promotes M2 phenotypic polarization, reverses the reduced neural stem cell in hippocampus, decreases the TNF-α level, and increases the IL-10 level in the blood. | [20][25] |
| BV-2 | γ-ray with 10 Gy (2.0 Gy/min) |
1.5, 6 h | FKN promotes microglial phagocytosis and M2 polarization, decreases TNF-α and IL-1β mRNA levels, and increases IL-10 mRNA levels. CX3CR1 knockdown reverses these effects. | [20][25] | ||
| PPARα | BV-2 | γ-ray with 10 Gy (4.0 Gy/min) |
1, 3, 7, 12, 24 h | PPARα activation prevents the increase in IL-1, and TNF-α mRNA levels, and COX-2 protein via inhibition of p65 translocation and jun phosphorylation. | [83][37] | |
| 129S1/SvImJ mouse | γ-ray with 10 Gy (3.33 Gy/min) |
whole brain | 1 week, 2 months | PPARα activation promotes newborn neuron survival and prevents microglial activation. PPARα knockout abolishes the neuroprotection of fenofibrate. | [201][99] | |
| Fischer 344 × Brown Norway rats | γ-ray with four fractions of 10 Gy (4 Gy/min) |
whole brain | 26, 29 weeks | PPARα activation prevents perirhinal cortex-dependent cognitive impairment without a decrease in microglial activation and an increase in immature neurons. | [78][31] | |
| PPARδ | BV-2 | γ-ray with 10 Gy (3.56 Gy/min) |
30 min; 7, 24 h | PPARδ activation downregulates ROS production, IL-1 and TNF-α expression, and COX-2 and MCP-1 proteins by inhibiting NF-κB and PKCα/MEK1/2/ERK1/2/AP pathways. | [18] | |
| C57BL/6J | γ-ray with 10 Gy (5 Gy/min) |
whole brain | 3 h; 1, 2 weeks | PPARδ activation prevents the increase in IL-1 gene expression and pERK protein but does not rescue neurogenesis and hippocampal-dependent cognitive impairment. | [203][100] | |
| PPARγ | Fischer 344 rat | γ-ray with nine fractions of 5 Gy (4.41 Gy/min) | whole brain | 50, 54 weeks | PPARγ activation prevents cognitive impairment. | [206][101] |
| Kv 1.3 | Balb/c mouse | ß-ray with 30 Gy (3 Gy/min) |
whole brain | 3, 14 days; 8 weeks | Kv 1.3 blockade prevents neuronal loss and increases activated microglial in hippocampus and cerebral cortex and improves spatial learning and cerebral cortex atrophy in mice. | [74][27] |
| BV-2 | ß-ray with 10 Gy (3 Gy/min) |
4, 12 h; 1, 2 days | Kv 1.3 blockade or knockdown decreases protein and mRNA level of TNF-α, IL-6, and COX-2 in microglia and inhibits apoptosis of co-cultured primary hippocampal neurons. | [74][27] | ||
| miR-124 | C57BL/6J mouse | γ-ray with 10 Gy (2.07 Gy/min) |
whole brain | 5 weeks | miR-124 overexpression prevents microglia activation and ameliorates cognitive impairment. | [231][102] |
| miR-741-3p | C57BL/6J mouse | ß-ray with 30 Gy (2.5 Gy/min) |
whole brain | 1, 6 weeks | miR-741-3p inhibition resists cognitive dysfunction, hippocampal neuronal injury, and microglia activation and decreases the expression level of IL-6 and TNF-a. | [229][103] |
| miR-122-5p | C57BL/6J mouse | ß-ray with 30 Gy (3 Gy/min) |
whole brain | 6 weeks, 48–50 days | miR-122-5p inhibition prevents cognitive impairment, neuronal damage, microglia activation, and production of TNF-a, IL-6, and IL-1ß in hippocampus. | [230][104] |
| BV-2 | ß-ray with 10 Gy | 8, 24 h | miR-122-5p inhibition alleviates the decrease in cell viability and increase in the release of TNF-a, IL-6, and IL-1ß in BV2; restores BV2 branching morphogenesis and phagocytosis; and reduces co-cultured SH-SY5Y cell apoptosis. | [230][104] | ||
| lncRNA ENSMUST00000130679 | BV-2 | X-ray with 10 Gy (2 Gy/min) |
1, 24 h | lncRNA ENSMUST00000130679 knockdown suppresses DDR; phosphorylation of p65, JNK, and p38; and release of TNF-a, IL-6, and IL-1ß in BV2. | [66][16] | |
| lncRNA ENSMUST00000190863 | BV-2 | X-ray with 10 Gy (2 Gy/min) |
1, 24 h | lncRNA ENSMUST00000190863 knockdown suppresses DDR, phosphorylation of p65, and release of TNF-a in BV2. | [66][16] | |
| hNSC-derived MV | athymic nude rats | X-ray with 10 Gy (1 Gy/min) |
whole brain | 4–7 weeks | MV transplantation into the bilateral hippocampus reduces the number of activated microglia in the hippocampus, neocortex (layer II/III), and amygdala; recovers the complexity of neuronal architecture; and ameliorates cognitive impairment. | [244][105] |
| 1 month | MV transplantation into the unilateral hippocampus reduces the number of activated microglia in the ipsilateral hippocampus; bilateral or unilateral transplantation increases GDNF and restores PSD-95 protein level in bilateral hippocampus; neither bilateral nor unilateral transplantation protects dendritic spine density. | [243][106] | ||||
| hNSC-derived EV | C57BL/6J mouse | γ-ray with 10 Gy (2.07 Gy/minute) |
whole brain | 5 weeks, 6 months | EV transplantation into the bilateral hippocampus prevents microglia activation in the hippocampus and ameliorates cognitive impairment. | [231][102] |
| ADMSC-Exos | Sprague–Dawley rats | γ-ray with 30 Gy (1.59 Gy/min) |
whole brain | 24 h; 3, 7 days | Tail vein injection pf ADMSC-Exos decreases the levels of caspase-3, MDA, 8-OHdG, TNF-α, IL-4, and SIRT1 and promotes recovery of SOD, CAT, IL-4, and IL-10 levels and suppresses microglial infiltration. | [73][26] |
| primary microglia | γ-ray with 30 Gy (3 MeV/min) |
24 h | Tail vein injection of ADMSC-Exos decreases the levels of caspase-3, MDA, 8-OHdG, TNF-α, IL-4, and SIRT1 and promotes the recovery of SOD, CAT, IL-4, and IL-10 levels and suppresses microglial activation. The above effects of ADMSC-Exos are inhibited by the SIRT-1 inhibitor EX527. | [73][26] |
References
- Ali, F.S.; Arevalo, O.; Zorofchian, S.; Patrizz, A.; Riascos, R.; Tandon, N.; Blanco, A.; Ballester, L.Y.; Esquenazi, Y. Cerebral Radiation Necrosis: Incidence, Pathogenesis, Diagnostic Challenges, and Future Opportunities. Curr. Oncol. Rep. 2019, 21, 66.
- Greene-Schloesser, D.; Robbins, M.E. Radiation-induced cognitive impairment--from bench to bedside. Neuro Oncol. 2012, 14 (Suppl. S4), iv37–iv44.
- Hladik, D.; Tapio, S. Effects of ionizing radiation on the mammalian brain. Mutat Res. Rev. Mutat Res. 2016, 770, 219–230.
- Brown, P.D.; Jaeckle, K.; Ballman, K.V.; Farace, E.; Cerhan, J.H.; Anderson, S.K.; Carrero, X.W.; Barker, F.G., 2nd; Deming, R.; Burri, S.H.; et al. Effect of Radiosurgery Alone vs Radiosurgery With Whole Brain Radiation Therapy on Cognitive Function in Patients With 1 to 3 Brain Metastases: A Randomized Clinical Trial. JAMA 2016, 316, 401–409.
- Chang, E.L.; Wefel, J.S.; Hess, K.R.; Allen, P.K.; Lang, F.F.; Kornguth, D.G.; Arbuckle, R.B.; Swint, J.M.; Shiu, A.S.; Maor, M.H.; et al. Neurocognition in patients with brain metastases treated with radiosurgery or radiosurgery plus whole-brain irradiation: A randomised controlled trial. Lancet Oncol. 2009, 10, 1037–1044.
- Turnquist, C.; Harris, B.T.; Harris, C.C. Radiation-induced brain injury: Current concepts and therapeutic strategies targeting neuroinflammation. Neurooncol. Adv. 2020, 2, vdaa057.
- Gutierrez-Quintana, R.; Walker, D.J.; Williams, K.J.; Forster, D.M.; Chalmers, A.J. Radiation-induced neuroinflammation: A potential protective role for poly(ADP-ribose) polymerase inhibitors? Neurooncol. Adv. 2022, 4, vdab190.
- Gondi, V.; Hermann, B.P.; Mehta, M.P.; Tome, W.A. Hippocampal dosimetry predicts neurocognitive function impairment after fractionated stereotactic radiotherapy for benign or low-grade adult brain tumors. Int J. Radiat. Oncol. Biol. Phys. 2012, 83, e487–e493.
- Florijn, M.A.; Sharfo, A.W.M.; Wiggenraad, R.G.J.; van Santvoort, J.P.C.; Petoukhova, A.L.; Hoogeman, M.S.; Mast, M.E.; Dirkx, M.L.P. Lower doses to hippocampi and other brain structures for skull-base meningiomas with intensity modulated proton therapy compared to photon therapy. Radiother. Oncol. 2020, 142, 147–153.
- Montay-Gruel, P.; Acharya, M.M.; Petersson, K.; Alikhani, L.; Yakkala, C.; Allen, B.D.; Ollivier, J.; Petit, B.; Jorge, P.G.; Syage, A.R.; et al. Long-term neurocognitive benefits of FLASH radiotherapy driven by reduced reactive oxygen species. Proc. Natl. Acad. Sci. USA 2019, 116, 10943–10951.
- Montay-Gruel, P.; Acharya, M.M.; Goncalves Jorge, P.; Petit, B.; Petridis, I.G.; Fuchs, P.; Leavitt, R.; Petersson, K.; Gondre, M.; Ollivier, J.; et al. Hypofractionated FLASH-RT as an Effective Treatment against Glioblastoma that Reduces Neurocognitive Side Effects in Mice. Clin. Cancer Res. 2021, 27, 775–784.
- Hwang, S.Y.; Jung, J.S.; Kim, T.H.; Lim, S.J.; Oh, E.S.; Kim, J.Y.; Ji, K.A.; Joe, E.H.; Cho, K.H.; Han, I.O. Ionizing radiation induces astrocyte gliosis through microglia activation. Neurobiol. Dis. 2006, 21, 457–467.
- Lumniczky, K.; Szatmari, T.; Safrany, G. Ionizing Radiation-Induced Immune and Inflammatory Reactions in the Brain. Front. Immunol. 2017, 8, 517.
- Yoshida, Y.; Sejimo, Y.; Kurachi, M.; Ishizaki, Y.; Nakano, T.; Takahashi, A. X-ray irradiation induces disruption of the blood-brain barrier with localized changes in claudin-5 and activation of microglia in the mouse brain. Neurochem. Int. 2018, 119, 199–206.
- Monje, M.L.; Mizumatsu, S.; Fike, J.R.; Palmer, T.D. Irradiation induces neural precursor-cell dysfunction. Nat. Med. 2002, 8, 955–962.
- Xu, A.; Li, R.; Ren, A.; Jian, H.; Huang, Z.; Zeng, Q.; Wang, B.; Zheng, J.; Chen, X.; Zheng, N.; et al. Regulatory coupling between long noncoding RNAs and senescence in irradiated microglia. J. Neuroinflamm. 2020, 17, 321.
- Jezkova, L.; Zadneprianetc, M.; Kulikova, E.; Smirnova, E.; Bulanova, T.; Depes, D.; Falkova, I.; Boreyko, A.; Krasavin, E.; Davidkova, M.; et al. Particles with similar LET values generate DNA breaks of different complexity and reparability: A high-resolution microscopy analysis of gammaH2AX/53BP1 foci. Nanoscale 2018, 10, 1162–1179.
- Schnegg, C.I.; Kooshki, M.; Hsu, F.C.; Sui, G.; Robbins, M.E. PPARdelta prevents radiation-induced proinflammatory responses in microglia via transrepression of NF-kappaB and inhibition of the PKCalpha/MEK1/2/ERK1/2/AP-1 pathway. Free Radic. Biol. Med. 2012, 52, 1734–1743.
- Xue, J.; Dong, J.H.; Huang, G.D.; Qu, X.F.; Wu, G.; Dong, X.R. NF-kappaB signaling modulates radiationinduced microglial activation. Oncol. Rep. 2014, 31, 2555–2560.
- Biber, K.; Neumann, H.; Inoue, K.; Boddeke, H.W. Neuronal ‘On’ and ‘Off’ signals control microglia. Trends Neurosci. 2007, 30, 596–602.
- Xu, P.; Xu, Y.; Hu, B.; Wang, J.; Pan, R.; Murugan, M.; Wu, L.J.; Tang, Y. Extracellular ATP enhances radiation-induced brain injury through microglial activation and paracrine signaling via P2X7 receptor. Brain Behav. Immun. 2015, 50, 87–100.
- Xu, L.; Huang, H.; Liu, T.; Yang, T.; Yi, X. Exposure to X-rays Causes Depression-like Behaviors in Mice via HMGB1-mediated Pyroptosis. Neuroscience 2022, 481, 99–110.
- Markarian, M.; Krattli, R.P., Jr.; Baddour, J.D.; Alikhani, L.; Giedzinski, E.; Usmani, M.T.; Agrawal, A.; Baulch, J.E.; Tenner, A.J.; Acharya, M.M. Glia-Selective Deletion of Complement C1q Prevents Radiation-Induced Cognitive Deficits and Neuroinflammation. Cancer Res. 2021, 81, 1732–1744.
- Boyd, A.; Byrne, S.; Middleton, R.J.; Banati, R.B.; Liu, G.J. Control of Neuroinflammation through Radiation-Induced Microglial Changes. Cells 2021, 10, 2381.
- Wang, J.; Pan, H.; Lin, Z.; Xiong, C.; Wei, C.; Li, H.; Tong, F.; Dong, X. Neuroprotective Effect of Fractalkine on Radia-tion-induced Brain Injury Through Promoting the M2 Polarization of Microglia. Mol. Neurobiol. 2021, 58, 1074–1087.
- Liu, M.; Yang, Y.; Zhao, B.; Yang, Y.; Wang, J.; Shen, K.; Yang, X.; Hu, D.; Zheng, G.; Han, J. Exosomes Derived From Adipose-Derived Mesenchymal Stem Cells Ameliorate Radiation-Induced Brain Injury by Activating the SIRT1 Pathway. Front. Cell Dev. Biol 2021, 9, 693782.
- Peng, Y.; Lu, K.; Li, Z.; Zhao, Y.; Wang, Y.; Hu, B.; Xu, P.; Shi, X.; Zhou, B.; Pennington, M.; et al. Blockade of Kv1.3 channels ameliorates radiation-induced brain injury. Neuro Oncol. 2014, 16, 528–539.
- Kalm, M.; Fukuda, A.; Fukuda, H.; Ohrfelt, A.; Lannering, B.; Bjork-Eriksson, T.; Blennow, K.; Marky, I.; Blomgren, K. Transient inflammation in neurogenic regions after irradiation of the developing brain. Radiat. Res. 2009, 171, 66–76.
- Chen, H.; Chong, Z.Z.; De Toledo, S.M.; Azzam, E.I.; Elkabes, S.; Souayah, N. Delayed activation of human microglial cells by high dose ionizing radiation. Brain Res. 2016, 1646, 193–198.
- Hong, J.H.; Chiang, C.S.; Campbell, I.L.; Sun, J.R.; Withers, H.R.; McBride, W.H. Induction of acute phase gene expression by brain irradiation. Int. J. Radiat. Oncol. Biol. Phys. 1995, 33, 619–626.
- Greene-Schloesser, D.; Payne, V.; Peiffer, A.M.; Hsu, F.C.; Riddle, D.R.; Zhao, W.; Chan, M.D.; Metheny-Barlow, L.; Robbins, M.E. The peroxisomal proliferator-activated receptor (PPAR) alpha agonist, fenofibrate, prevents fractionated whole-brain irradiation-induced cognitive impairment. Radiat. Res. 2014, 181, 33–44.
- Jenrow, K.A.; Brown, S.L.; Lapanowski, K.; Naei, H.; Kolozsvary, A.; Kim, J.H. Selective inhibition of microglia-mediated neuroinflammation mitigates radiation-induced cognitive impairment. Radiat. Res. 2013, 179, 549–556.
- Casciati, A.; Dobos, K.; Antonelli, F.; Benedek, A.; Kempf, S.J.; Belles, M.; Balogh, A.; Tanori, M.; Heredia, L.; Atkinson, M.J.; et al. Age-related effects of X-ray irradiation on mouse hippocampus. Oncotarget 2016, 7, 28040–28058.
- Bachiller, S.; Jimenez-Ferrer, I.; Paulus, A.; Yang, Y.; Swanberg, M.; Deierborg, T.; Boza-Serrano, A. Microglia in Neurological Diseases: A Road Map to Brain-Disease Dependent-Inflammatory Response. Front. Cell. Neurosci. 2018, 12, 488.
- Betlazar, C.; Middleton, R.J.; Banati, R.B.; Liu, G.J. The impact of high and low dose ionising radiation on the central nervous system. Redox Biol. 2016, 9, 144–156.
- Claude, J.; Linnartz-Gerlach, B.; Kudin, A.P.; Kunz, W.S.; Neumann, H. Microglial CD33-related Siglec-E inhibits neurotoxicity by preventing the phagocytosis-associated oxidative burst. J. Neurosci. 2013, 33, 18270–18276.
- Ramanan, S.; Kooshki, M.; Zhao, W.; Hsu, F.C.; Robbins, M.E. PPARalpha ligands inhibit radiation-induced microglial inflammatory responses by negatively regulating NF-kappaB and AP-1 pathways. Free Radic. Biol. Med. 2008, 45, 1695–1704.
- Leach, J.K.; Van Tuyle, G.; Lin, P.S.; Schmidt-Ullrich, R.; Mikkelsen, R.B. Ionizing radiation-induced, mitochondria-dependent generation of reactive oxygen/nitrogen. Cancer Res. 2001, 61, 3894–3901.
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 2020, 9, 743.
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947.
- Cho, H.J.; Lee, W.H.; Hwang, O.M.H.; Sonntag, W.E.; Lee, Y.W. Role of NADPH oxidase in radiation-induced pro-oxidative and pro-inflammatory pathways in mouse brain. Int J. Radiat. Biol. 2017, 93, 1257–1266.
- Deng, Z.; Sui, G.; Rosa, P.M.; Zhao, W. Radiation-induced c-Jun activation depends on MEK1-ERK1/2 signaling pathway in microglial cells. PLoS ONE 2012, 7, e36739.
- Han, J.E.; Choi, J.W. Control of JNK for an activation of NADPH oxidase in LPS-stimulated BV2 microglia. Arch. Pharm Res. 2012, 35, 709–715.
- Betlazar, C.; Middleton, R.J.; Howell, N.; Storer, B.; Davis, E.; Davies, J.; Banati, R.; Liu, G.J. Mitochondrial Translocator Protein (TSPO) Expression in the Brain After Whole Body Gamma Irradiation. Front. Cell Dev. Biol. 2021, 9, 715444.
- Anholt, R.R.; Pedersen, P.L.; De Souza, E.B.; Snyder, S.H. The peripheral-type benzodiazepine receptor. Localization to the mitochondrial outer membrane. J. Biol. Chem. 1986, 261, 576–583.
- Choi, J.; Ifuku, M.; Noda, M.; Guilarte, T.R. Translocator protein (18 kDa)/peripheral benzodiazepine receptor specific ligands induce microglia functions consistent with an activated state. Glia 2011, 59, 219–230.
- Wolf, A.; Herb, M.; Schramm, M.; Langmann, T. The TSPO-NOX1 axis controls phagocyte-triggered pathological angiogenesis in the eye. Nat. Commun. 2020, 11, 2709.
- Loth, M.K.; Guariglia, S.R.; Re, D.B.; Perez, J.; de Paiva, V.N.; Dziedzic, J.L.; Chambers, J.W.; Azzam, D.J.; Guilarte, T.R. A Novel Interaction of Translocator Protein 18 kDa (TSPO) with NADPH Oxidase in Microglia. Mol. Neurobiol. 2020, 57, 4467–4487.
- Lee, J.W.; Kim, L.E.; Shim, H.J.; Kim, E.K.; Hwang, W.C.; Min, D.S.; Yu, S.W. A translocator protein 18 kDa ligand, Ro5-4864, inhibits ATP-induced NLRP3 inflammasome activation. Biochem. Biophys. Res. Commun. 2016, 474, 587–593.
- Batarseh, A.; Li, J.; Papadopoulos, V. Protein kinase C epsilon regulation of translocator protein (18 kDa) Tspo gene expression is mediated through a MAPK pathway targeting STAT3 and c-Jun transcription factors. Biochemistry 2010, 49, 4766–4778.
- Zhao, Y.Y.; Yu, J.Z.; Li, Q.Y.; Ma, C.G.; Lu, C.Z.; Xiao, B.G. TSPO-specific ligand vinpocetine exerts a neuroprotective effect by suppressing microglial inflammation. Neuron Glia Biol. 2011, 7, 187–197.
- Choi, H.B.; Khoo, C.; Ryu, J.K.; van Breemen, E.; Kim, S.U.; McLarnon, J.G. Inhibition of lipopolysaccharide-induced cyclooxygenase-2, tumor necrosis factor-alpha and i responses in human microglia by the peripheral benzodiazepine receptor ligand PK11195. J. Neurochem. 2002, 83, 546–555.
- Lee, J.W.; Nam, H.; Yu, S.W. Systematic Analysis of Translocator Protein 18 kDa (TSPO) Ligands on Toll-like Receptors-mediated Pro-inflammatory Responses in Microglia and Astrocytes. Exp. Neurobiol. 2016, 25, 262–268.
- Betlazar, C.; Middleton, R.J.; Banati, R.; Liu, G.J. The Translocator Protein (TSPO) in Mitochondrial Bioenergetics and Immune Processes. Cells. 2020, 9, 512.
- Azrad, M.; Zeineh, N.; Weizman, A.; Veenman, L.; Gavish, M. The TSPO Ligands 2-Cl-MGV-1, MGV-1, and PK11195 Differentially Suppress the Inflammatory Response of BV-2 Microglial Cell to LPS. Int. J. Mol. Sci. 2019, 20, 594.
- Monga, S.; Nagler, R.; Amara, R.; Weizman, A.; Gavish, M. Inhibitory Effects of the Two Novel TSPO Ligands 2-Cl-MGV-1 and MGV-1 on LPS-induced Microglial Activation. Cells. 2019, 8, 486.
- Dringen, R. Oxidative and antioxidative potential of brain microglial cells. Antioxid. Redox Signal. 2005, 7, 1223–1233.
- Ismail, A.F.; El-Sonbaty, S.M. Fermentation enhances Ginkgo biloba protective role on gamma-irradiation induced neuroinflammatory gene expression and stress hormones in rat brain. J. Photochem. Photobiol. B 2016, 158, 154–163.
- Fishman, K.; Baure, J.; Zou, Y.; Huang, T.T.; Andres-Mach, M.; Rola, R.; Suarez, T.; Acharya, M.; Limoli, C.L.; Lamborn, K.R.; et al. Radiation-induced reductions in neurogenesis are ameliorated in mice deficient in CuZnSOD or MnSOD. Free Radic. Biol. Med. 2009, 47, 1459–1467.
- Choi, S.H.; Aid, S.; Kim, H.W.; Jackson, S.H.; Bosetti, F. Inhibition of NADPH oxidase promotes alternative and anti-inflammatory microglial activation during neuroinflammation. J. Neurochem. 2012, 120, 292–301.
- Bhat, S.A.; Sood, A.; Shukla, R.; Hanif, K. AT2R Activation Prevents Microglia Pro-inflammatory Activation in a NOX-Dependent Manner: Inhibition of PKC Activation and p47(phox) Phosphorylation by PP2A. Mol. Neurobiol. 2019, 56, 3005–3023.
- Vilhardt, F.; Haslund-Vinding, J.; Jaquet, V.; McBean, G. Microglia antioxidant systems and redox signalling. Br. J. Pharmacol. 2017, 174, 1719–1732.
- Hamada, N.; Matsumoto, H.; Hara, T.; Kobayashi, Y. Intercellular and intracellular signaling pathways mediating ionizing radiation-induced bystander effects. J. Radiat. Res. 2007, 48, 87–95.
- Haslund-Vinding, J.; McBean, G.; Jaquet, V.; Vilhardt, F. NADPH oxidases in oxidant production by microglia: Activating receptors, pharmacology and association with disease. Br. J. Pharmacol. 2017, 174, 1733–1749.
- Zhang, F.; Qian, L.; Flood, P.M.; Shi, J.S.; Hong, J.S.; Gao, H.M. Inhibition of IkappaB kinase-beta protects dopamine neurons against lipopolysaccharide-induced neurotoxicity. J. Pharmacol. Exp. Ther. 2010, 333, 822–833.
- Rojo, A.I.; McBean, G.; Cindric, M.; Egea, J.; Lopez, M.G.; Rada, P.; Zarkovic, N.; Cuadrado, A. Redox control of microglial function: Molecular mechanisms and functional significance. Antioxid Redox Signal. 2014, 21, 1766–1801.
- Park, J.; Min, J.S.; Kim, B.; Chae, U.B.; Yun, J.W.; Choi, M.S.; Kong, I.K.; Chang, K.T.; Lee, D.S. Mitochondrial ROS govern the LPS-induced pro-inflammatory response in microglia cells by regulating MAPK and NF-kappaB pathways. Neurosci. Lett. 2015, 584, 191–196.
- Mehrabadi, A.R.; Korolainen, M.A.; Odero, G.; Miller, D.W.; Kauppinen, T.M. Poly(ADP-ribose) polymerase-1 regulates microglia mediated decrease of endothelial tight junction integrity. Neurochem. Int. 2017, 108, 266–271.
- Haruwaka, K.; Ikegami, A.; Tachibana, Y.; Ohno, N.; Konishi, H.; Hashimoto, A.; Matsumoto, M.; Kato, D.; Ono, R.; Kiyama, H.; et al. Dual microglia effects on blood brain barrier permeability induced by systemic inflammation. Nat. Commun. 2019, 10, 5816.
- Yuan, H.; Gaber, M.W.; McColgan, T.; Naimark, M.D.; Kiani, M.F.; Merchant, T.E. Radiation-induced permeability and leukocyte adhesion in the rat blood-brain barrier: Modulation with anti-ICAM-1 antibodies. Brain Res. 2003, 969, 59–69.
- Mildner, A.; Schmidt, H.; Nitsche, M.; Merkler, D.; Hanisch, U.K.; Mack, M.; Heikenwalder, M.; Bruck, W.; Priller, J.; Prinz, M. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat. Neurosci. 2007, 10, 1544–1553.
- Morganti, J.M.; Jopson, T.D.; Liu, S.; Gupta, N.; Rosi, S. Cranial irradiation alters the brain’s microenvironment and permits CCR2+ macrophage infiltration. PLoS ONE 2014, 9, e93650.
- Yuan, H.; Gaber, M.W.; Boyd, K.; Wilson, C.M.; Kiani, M.F.; Merchant, T.E. Effects of fractionated radiation on the brain vasculature in a murine model: Blood-brain barrier permeability, astrocyte proliferation, and ultrastructural changes. Int. J. Radiat. Oncol. Biol. Phys. 2006, 66, 860–866.
- Allen, B.D.; Apodaca, L.A.; Syage, A.R.; Markarian, M.; Baddour, A.A.D.; Minasyan, H.; Alikhani, L.; Lu, C.; West, B.L.; Giedzinski, E.; et al. Attenuation of neuroinflammation reverses Adriamycin-induced cognitive impairments. Acta Neuropathol. Commun. 2019, 7, 186.
- Wang, J.J.; Tong, F.; Lin, Z.Y.; Dong, X.R. The Effects of Vascular Endothelial Cells on Regulating Post-Irradiation Microglia Phenotype in Irradiation-Induced Brain Injury. J. Thorac. Oncol. 2021, 16, 71.
- Khan, S.Y.; Awad, E.M.; Oszwald, A.; Mayr, M.; Yin, X.; Waltenberger, B.; Stuppner, H.; Lipovac, M.; Uhrin, P.; Breuss, J.M. Premature senescence of endothelial cells upon chronic exposure to TNFalpha can be prevented by N-acetyl cysteine and plumericin. Sci. Rep. 2017, 7, 39501.
- Kyrkanides, S.; Olschowka, J.A.; Williams, J.P.; Hansen, J.T.; O’Banion, M.K. TNF alpha and IL-1beta mediate intercellular adhesion molecule-1 induction via microglia-astrocyte interaction in CNS radiation injury. J. Neuroimmunol. 1999, 95, 95–106.
- Jang, C.; Kim, J.; Kwon, Y.; Jo, S.A. Telmisartan Inhibits TNFalpha-Induced Leukocyte Adhesion by Blocking ICAM-1 Expression in Astroglial Cells but Not in Endothelial Cells. Biomol. Ther. 2020, 28, 423–430.
- Wilson, C.M.; Gaber, M.W.; Sabek, O.M.; Zawaski, J.A.; Merchant, T.E. Radiation-induced astrogliosis and blood-brain barrier damage can be abrogated using anti-TNF treatment. Int. J. Radiat. Oncol. Biol. Phys. 2009, 74, 934–941.
- Ronaldson, P.T.; Davis, T.P. Regulation of blood-brain barrier integrity by microglia in health and disease: A therapeutic opportunity. J. Cereb. Blood Flow Metab. 2020, 40, S6–S24.
- Moravan, M.J.; Olschowka, J.A.; Williams, J.P.; O’Banion, M.K. Cranial irradiation leads to acute and persistent neuroinflammation with delayed increases in T-cell infiltration and CD11c expression in C57BL/6 mouse brain. Radiat. Res. 2011, 176, 459–473.
- Burrell, K.; Hill, R.P.; Zadeh, G. High-resolution in-vivo analysis of normal brain response to cranial irradiation. PLoS ONE 2012, 7, e38366.
- Moravan, M.J.; Olschowka, J.A.; Williams, J.P.; O’Banion, M.K. Brain radiation injury leads to a dose- and time-dependent recruitment of peripheral myeloid cells that depends on CCR2 signaling. J. Neuroinflamm. 2016, 13, 30.
- Belarbi, K.; Jopson, T.; Arellano, C.; Fike, J.R.; Rosi, S. CCR2 deficiency prevents neuronal dysfunction and cognitive impairments induced by cranial irradiation. Cancer Res. 2013, 73, 1201–1210.
- Osman, A.M.; Sun, Y.; Burns, T.C.; He, L.; Kee, N.; Oliva-Vilarnau, N.; Alevyzaki, A.; Zhou, K.; Louhivuori, L.; Uhlen, P.; et al. Radiation Triggers a Dynamic Sequence of Transient Microglial Alterations in Juvenile Brain. Cell Rep. 2020, 31, 107699.
- Whitelaw, B.S.; Tanny, S.; Johnston, C.J.; Majewska, A.K.; O’Banion, M.K.; Marples, B. In Vivo Imaging of the Microglial Landscape after Whole Brain Radiation Therapy. Int. J. Radiat. Oncol. Biol. Phys. 2021, 111, 1066–1071.
- Lee, S.W.; Haditsch, U.; Cord, B.J.; Guzman, R.; Kim, S.J.; Boettcher, C.; Priller, J.; Ormerod, B.K.; Palmer, T.D. Absence of CCL2 is sufficient to restore hippocampal neurogenesis following cranial irradiation. Brain Behav. Immun. 2013, 30, 33–44.
- Acharya, M.M.; Patel, N.H.; Craver, B.M.; Tran, K.K.; Giedzinski, E.; Tseng, B.P.; Parihar, V.K.; Limoli, C.L. Consequences of low dose ionizing radiation exposure on the hippocampal microenvironment. PLoS ONE 2015, 10, e0128316.
- Raber, J.; Allen, A.R.; Rosi, S.; Sharma, S.; Dayger, C.; Davis, M.J.; Fike, J.R. Effects of (56)Fe radiation on hippocampal function in mice deficient in chemokine receptor 2 (CCR2). Behav. Brain Res. 2013, 246, 69–75.
- Dietrich, J.; Baryawno, N.; Nayyar, N.; Valtis, Y.K.; Yang, B.; Ly, I.; Besnard, A.; Severe, N.; Gustafsson, K.U.; Andronesi, O.C.; et al. Bone marrow drives central nervous system regeneration after radiation injury. J. Clin. Investig. 2018, 128, 281–293.
- Acharya, M.M.; Green, K.N.; Allen, B.D.; Najafi, A.R.; Syage, A.; Minasyan, H.; Le, M.T.; Kawashita, T.; Giedzinski, E.; Parihar, V.K.; et al. Elimination of microglia improves cognitive function following cranial irradiation. Sci. Rep. 2016, 6, 31545.
- Allen, B.D.; Syage, A.R.; Maroso, M.; Baddour, A.A.D.; Luong, V.; Minasyan, H.; Giedzinski, E.; West, B.L.; Soltesz, I.; Limoli, C.L.; et al. Mitigation of helium irradiation-induced brain injury by microglia depletion. J. Neuroinflamm. 2020, 17, 159.
- Krukowski, K.; Feng, X.; Paladini, M.S.; Chou, A.; Sacramento, K.; Grue, K.; Riparip, L.K.; Jones, T.; Campbell-Beachler, M.; Nelson, G.; et al. Temporary microglia-depletion after cosmic radiation modifies phagocytic activity and prevents cognitive deficits. Sci. Rep. 2018, 8, 7857.
- Feng, X.; Jopson, T.D.; Paladini, M.S.; Liu, S.; West, B.L.; Gupta, N.; Rosi, S. Colony-stimulating factor 1 receptor blockade prevents fractionated whole-brain irradiation-induced memory deficits. J. Neuroinflamm. 2016, 13, 215.
- Kalm, M.; Andreasson, U.; Bjork-Eriksson, T.; Zetterberg, H.; Pekny, M.; Blennow, K.; Pekna, M.; Blomgren, K. C3 deficiency ameliorates the negative effects of irradiation of the young brain on hippocampal development and learning. Oncotarget 2016, 7, 19382–19394.
- Hinkle, J.J.; Olschowka, J.A.; Williams, J.P.; O’Banion, M.K. Pharmacologic manipulation of complement receptor 3 prevents dendritic spine loss and cognitive impairment after acute cranial radiation. bioRxiv 2020.
- Hinkle, J.J.; Olschowka, J.A.; Love, T.M.; Williams, J.P.; O’Banion, M.K. Cranial irradiation mediated spine loss is sex-specific and complement receptor-3 dependent in male mice. Sci. Rep. 2019, 9, 18899.
- Xu, Y.; Hu, W.; Liu, Y.; Xu, P.; Li, Z.; Wu, R.; Shi, X.; Tang, Y. P2Y6 Receptor-Mediated Microglial Phagocytosis in Radiation-Induced Brain Injury. Mol. Neurobiol. 2016, 53, 3552–3564.
- Ramanan, S.; Kooshki, M.; Zhao, W.; Hsu, F.C.; Riddle, D.R.; Robbins, M.E. The PPARalpha agonist fenofibrate preserves hippocampal neurogenesis and inhibits microglial activation after whole-brain irradiation. Int. J. Radiat. Oncol. Biol. Phys. 2009, 75, 870–877.
- Schnegg, C.I.; Greene-Schloesser, D.; Kooshki, M.; Payne, V.S.; Hsu, F.C.; Robbins, M.E. The PPARδ agonist GW0742 inhibits neuroinflammation, but does not restore neurogenesis or prevent early delayed hippocampal-dependent cognitive impairment after whole-brain irradiation. Free Radic. Biol. Med. 2013, 61, 1–9.
- Zhao, W.; Payne, V.; Tommasi, E.; Diz, D.I.; Hsu, F.C.; Robbins, M.E. Administration of the peroxisomal proliferator-activated receptor gamma agonist pioglitazone during fractionated brain irradiation prevents radiation-induced cognitive impairment. Int. J. Radiat. Oncol. Biol. Phys. 2007, 67, 6–9.
- Leavitt, R.J.; Acharya, M.M.; Baulch, J.E.; Limoli, C.L. Extracellular Vesicle-Derived miR-124 Resolves Radiation-Induced Brain Injury. Cancer Res. 2020, 80, 4266–4277.
- Ou, M.; Fan, W.; Sun, F.; Li, M.; Lin, M.; Yu, Y.; Liang, S.; Liao, H.; Jie, W.; Cai, Y.; et al. Nasal Delivery of AntagomiR-741 Protects Against the Radiation-Induced Brain Injury in Mice. Radiat. Res. 2021, 195, 355–365.
- Zhou, H.; Sun, F.; Ou, M.; Zhang, Y.; Lin, M.; Song, L.; Yu, Y.; Liao, H.; Fan, W.; Xing, H.; et al. Prior nasal delivery of antagomiR-122 prevents radiation-induced brain injury. Mol. Ther 2021, 29, 3465–3483.
- Baulch, J.E.; Acharya, M.M.; Allen, B.D.; Ru, N.; Chmielewski, N.N.; Martirosian, V.; Giedzinski, E.; Syage, A.; Park, A.L.; Benke, S.N.; et al. Cranial grafting of stem cell-derived microvesicles improves cognition and reduces neuropathology in the irradiated brain. Proc. Natl. Acad. Sci. USA 2016, 113, 4836–4841.
- Smith, S.M.; Giedzinski, E.; Angulo, M.C.; Lui, T.; Lu, C.; Park, A.L.; Tang, S.; Martirosian, V.; Ru, N.; Chmielewski, N.N.; et al. Functional equivalence of stem cell and stem cell-derived extracellular vesicle transplantation to repair the irradiated brain. Stem Cells Transl. Med. 2020, 9, 93–105.
More