Multiple sclerosis (MS) is an inflammatory demyelinating autoimmune disease of the central nervous system (CNS) that typically manifests with episodes of new/recurrent neurological symptoms (i.e., relapses) followed by either complete remission or residual disability (relapsing–remitting, RR, course).
Recent evidence of effectiveness of B cell-depleting monoclonal antibodies (mAbs) in MS prompted a partial revisitation of the pathogenetic paradigm of the disease, which has been, so far, considered a T cell-mediated autoimmune disorder. Although mechanisms underlying the efficacy of B cell-depleting mAbs in MS are not fully elucidated, they likely involve the impairment of pleiotropic B cell functions different from antibody secretion, such as their role as antigen-presenting cells. A potential impact of B cell-depleting mAbs on inflammation compartmentalized within the central nervous system was also suggested, but little is known about the mechanism underlying this latter phenomenon.
- multiple sclerosis
- B cell-depleting therapy
- monoclonal antibody
1. Introduction
2. Insights into MS Pathogenesis: Onset of Autoimmunity
MS was traditionally considered a T-cell-mediated autoimmune disorder, based on preclinical data from animal models of the disease (experimental autoimmune encephalomyelitis—EAE) and evidence for T-cell infiltration in inflammatory lesions and normal-appearing white matter of autoptic and biopsy CNS specimens from affected individuals, with an association between CD8+ T cells number and axonal damage [5][6][7][8][9][10][11][12][5,6,7,8,9,10,11,12]. Furthermore, the identification of expanded T-cell clones in the brain parenchyma, cerebrospinal fluid (CSF), and peripheral blood of MS patients detected using T-cell receptor (TCR) analyses reinforced the hypothesis that inflammatory infiltrates were constituted by pathogenetic expanded T-cell clones reactive to myelin antigens [13][14][15][16][13,14,15,16]. Circulating CD4+ T cells from MS patients were indeed demonstrated to recognise myelin basic protein (MBP), proteolipid protein (PLP), and myelin oligodendrocyte glycoprotein (MOG), even if the same phenomenon was also observed in healthy individuals; evidence regarding potential differences between these groups in frequency and avidity of cell interactions is conflicting [17][18][17,18]. A contribution of B cells to MS pathogenesis was also suggested by preclinical and clinical evidence, and their role was recently re-evaluated with the observation of a remarkable therapeutic effect of B-cell-depleting strategies [19]. Innate immunity cells, including CNS-resident microglia, contribute to MS pathogenesis, and neurodegenerative phenomena possibly, at least in part, independent of inflammation play a role in advanced disease [3]. Although MS aetiology is unknown, several environmental and genetic risk factors were identified [20][21][22][20,21,22]. Class II major histocompatibility complex (MHCII) represents the major genetic risk factor, accounting for 20–30% of individual genetic susceptibility [22][23][22,23]. MHCII genes encode membrane glycoproteins that are expressed by professional antigen-presenting cells (APC), such as dendritic cells, macrophages, and B cells [24]. The MHCII complex plays a key role in the development of both primary and secondary T CD4-mediated immune response as it presents in its context small peptide antigens processed by professional APC to MHC-restricted T cells [25]. In addition to MHC, several other genes were associated with the risk of developing MS on the basis of a complex genetic background, as confirmed by genome-wide association studies (GWAS) that uncovered more than 200 genetic susceptibility variants which could jointly account for ~48% of the estimated heritability for MS [26]. Enrichment for MS susceptibility loci was mostly related to genes involved in immune system function and regulation, and it was apparent in many different immune cell types and tissues, including microglia, highlighting, overall, the relevance of adaptive and innate immune cells in MS pathogenesis.2.1. Primary Autoimmune Response in the Peripheral Compartment
The mechanisms which trigger MS are still debated and two plausibly complementary pathogenetic models were proposed to explain the initiation of the autoimmune response, suggesting that the primum movens might take place either in the periphery (“outside-in” model) or within the CNS (“inside-out” model) [27]. The “outside-in” (or CNS-extrinsic/peripheral) model resembles the pathogenetic mechanism underlying EAE in which the disease is induced by external immunisation obtained through the inoculation of myelin-specific antigens in combination with an adjuvant [28]. According to this model, activation and expansion of CNS antigen-specific CD4+ T cells occur in the periphery and may be induced by an encounter with exogenous antigens sharing structural motifs with myelin antigens, hence capable of eliciting an autoimmune response based on molecular mimicry [29][30][29,30]. Several infective agents were suggested as potential exogenous triggers of the autoimmune reaction, with Epstein–Barr virus (EBV) the most plausible candidate [31][32][31,32]. On the other hand, the “inside-out” model suggests that the autoimmune reaction is triggered by an “internal event” occurring within the CNS and generating myelin debris. CNS-derived soluble antigens may then be drained via lymphatic pathways to peripheral lymphoid organs, where they might be presented to T and B cells eliciting the autoimmune reaction [33][34][33,34]. Different hypotheses suggest that the CNS-intrinsic event might be triggered by a CNS viral infection or by primary neurodegeneration [11].2.2. Secondary Autoimmune Response in the Central Nervous System
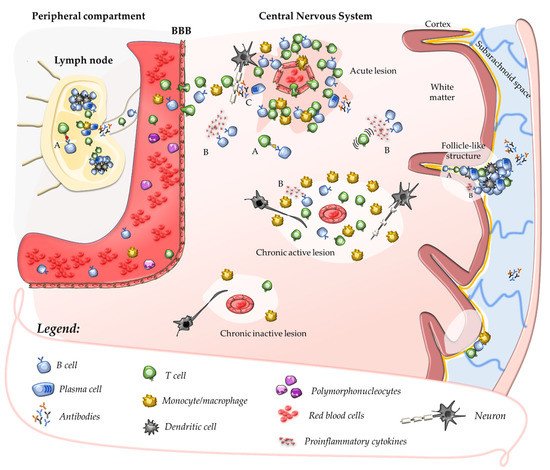
Once their priming has occurred, autoreactive T cells might be further expanded in peripheral lymphoid tissues after the encounter with their cognate antigen [22][35][22,35]. In the absence of neuroinflammation, T cells patrol the CNS crossing the BBB at the level of CNS post-capillary venules and reaching perivascular or subarachnoid spaces [36]. In such areas, putative cross-reactive T-cell clones may be further activated by CNS antigens (sharing structural motifs with their cognate antigen) which, after being drained from the parenchyma, are processed and presented by tissue-resident APCs. This mechanism promotes a secondary immune response within the CNS characterised by cell proliferation, recruitment of pro-inflammatory cells, and formation of perivascular cuffs around post-capillary and pial venules [37]. Upon activation, adaptive immune cells cross the glia limitans and infiltrate the CNS parenchyma where they produce pro-inflammatory cytokines and chemokines that determine a breakdown of the BBB. This causes further recruitment of adaptive and innate immune cells, which ultimately promote the formation of demyelinating lesions and tissue injury (Figure 1) [37][38][37,38]. Further damage to CNS tissue might derive from uncovering and release in the extracellular space of several autoantigens which, in turn, might enhance the autoimmune response in a mechanism defined as epitope spreading [39].