Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 3 by Dean Liu and Version 2 by Dean Liu.
Th17 cells are the main source of the proinflammatory cytokine IL-17; however, the receptor of this cytokine (IL-17R) is distributed ubiquitously. IL-17 (IL-17A) is a member of the IL-17 cytokine family consisting of IL-17A–F (IL-17E is also known as IL-25) and directly links inflammatory responses and T-cell activation.
- Th17 lymphocytes
- dendritic cells
- breast cancer
1. Introduction
After activation, CD4+ T lymphocytes, which are the central regulatory cells of innate and adaptive immunity, differentiate into various T helper (Th) subsets to ensure homeostasis. Among these subsets, the well-known ones are Th1 cells producing interferon-γ (IFN-γ) and Th2 producing interleukin (IL) 4. The other identified Th subset is Th17 producing IL-17. CD4+ T cells also differentiate into T regulatory cells (Treg) expressing forkhead box P3 (FOXP3) [1] (Figure 1).
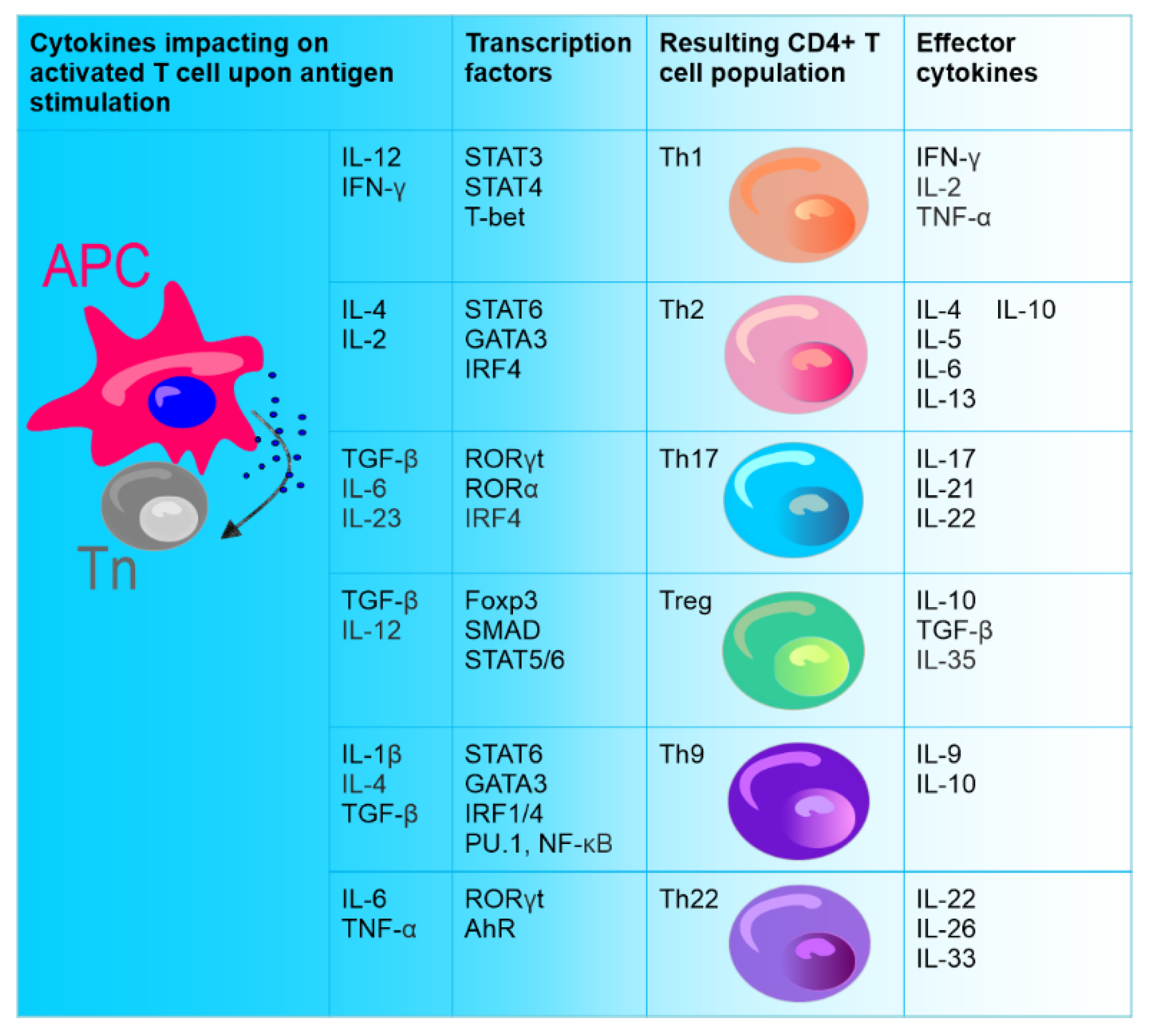
Figure 1. Differentiation of naïve CD4+ T cells (Tn). Naive CD4+ T cells (stem cell-like cells), under the influence of different cytokines secreted upon direct contact with antigen-presenting cell (APC), can differentiate into various types of effector cells: Th1, Th2, Th9, Th17, Th22, and Treg. CD4+ T cell subsets are defined by the production of specific cytokines and the expression of specific transcription factors.
As transforming growth factor β (TGF-β) plays an important role in the differentiation of both Th17 and Treg cells (Figure 1), IL-6 counteracts the differentiation of Treg cells upon TGF-β and directs the differentiation of Th17 [2][3]. It has been shown that IL-6 upregulates the expression of IL-21 by activating signal transducer and activator of transcription 3 (STAT3), causing an increase in the expression of retinoid-acid receptor-related orphan nuclear receptor (ROR) γt, RORα, and IL-23R and ultimately promoting the complete differentiation of Th17 cells. On the other hand, STAT3 deficiency impaired RORγt expression and elevated the expression of T-bet (member of T-box family transcription factors) and FOXP3 [4]. RORγt belongs to the retinol family and regulates the differentiation of Th17 cells, while RORα promotes the differentiation of these cells. RORγt and RORα can synergistically induce Th17 differentiation [5]. IL-21 produced by Th17 cells stimulates their autocrine formation [6]. IL-1 receptor type 1 (Il1r1) gene is also the promoter of Th17 cell differentiation [7]. Moreover, Th17 cells express CD39 and CD73 ectonucleotidases, leading to the release of adenosine and the suppression of effector T cells.
As Chalmin et al. have shown, during the differentiation of Th17 cells, the ectonucleotidase expression is transcriptionally regulated by IL-6 (STAT3 activation) and by TGF-β-mediated downregulation of zinc finger protein growth factor independent-1 (Gfi-1). The expression of CD39 ectonucleotidase in the case of Th17 cells determines their immunosuppressive nature in cancer [8].
In vitro induction of Th17 cells can be achieved directly with the use of anti-CD3/CD28 antibodies and cytokines. However, in vivo priming of Th17 cells requires dendritic cells (DCs) that present antigen, provide costimulatory signals, and help in the synthesis of IL-1, IL-6, TGF-β, tumor necrosis factor (TNF) α, and IL-23 cytokines [9][10][11]. It is also known that fibroblasts support IL-23 secretion from DCs that are preactivated by lipopolysaccharide compared to DCs activated by lipopolysaccharide alone. It is realized via a complex feedback-loop mechanism involving IL-1β/TNF-α (from activated DCs), which stimulates prostaglandin E2 (PGE2) production by fibroblasts. PGE2, in turn, acts on activated DCs and increases the release of IL-23 from these cells. Furthermore, compared to DCs alone, fibroblast-stimulated DCs performed better in promoting the expansion of Th17 cells in a cyclooxygenase (COX)-2-, IL-23-dependent manner [12] (Figure 2). A recent review by Pastor-Fernandez et al. broadly described the role of IL-23 in the differentiation of Th17 cells [13]. The role of IL-33 in maintaining the balance between Treg and Th17 cells has also been emphasized. DCs matured upon IL-33 inhibited the differentiation of CD4+ Treg cells in favor of Th17, which was realized through IL-6 signaling [14].
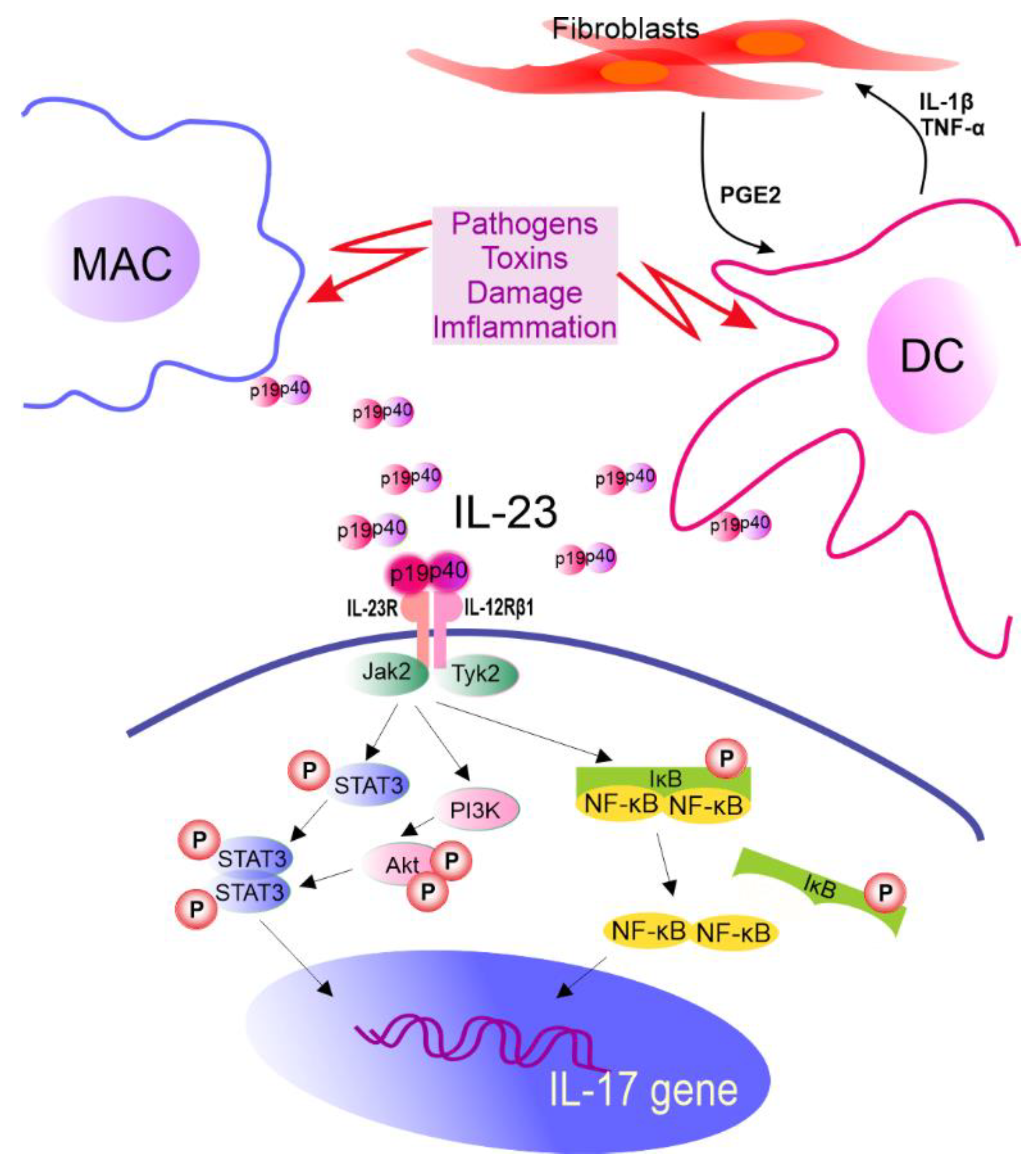
Figure 2. The role of IL-23 in IL-17 gene expression. IL-23R pairs with IL-12Rβ1 forming IL-23R complex required for IL-23 signaling. This receptor is constitutively associated with Janus kinase 2 (Jak2) and Tyrosine kinase 2 (Tyk2) which are activated after ligand biding, leading to STAT3 phosphorylation (P). Other molecules in IL-23 signaling cascade are also identified. MAC—macrophages, DC—dendritic cells, IL—interleukin, PGE2—prostaglandin E2, TNF-α—tumor necrosis factor α, STAT3—signal transducer and activator of transcription 3, PI3K—phosphoinositide 3-kinase, Akt—serine/threonine-protein kinase, NF-κB—nuclear factor kappa-light-chain-enhancer of activated B cells, IκB—NF-κB inhibitor, and p19 and p40—subunits of IL-23.
Recent studies suggest that osteopontin (OPN) is also required for DCs to induce Th17 differentiation [15] and IL-17 production [16]. In addition, studies on acute coronary syndrome showed that OPN is involved in inflammation through its direct effect on IL-17-producing cells [17]. Moreover, the direct effect of OPN on the differentiation of Th17 cells is exerted through interaction of these cells with its receptors [18]. OPN contains an Arg-Gly-Asp (RGD) sequence, which is common to many extracellular matrix proteins and mediates the interaction of OPN with multiple integrins such as αvβ1, αvβ3, αvβ5, and α5β1 [19]. CD44, which is another important receptor of OPN, is involved in T cell activation [20]. Studies have shown that blocking of CD44 and, to a lesser extent, β1 integrin subunit (CD29), resulted in a significant reduction in Th17 cell differentiation, while the addition of a CD51 (integrin αv)-blocking antibody did not result in such effect, indicating that the effect of OPN was mediated through CD44 and CD29. Furthermore, the production of IL-17 from OPN-stimulated CD4+ T cells was inhibited by CD44 or CD29 antibodies in a dose-dependent manner [21]. Other studies have also pointed out the role of CD61 (β3 integrin), another OPN receptor, in Th17 cell differentiation [18].
Estradiol (E2) is also a factor that affects Th17 cells. Th17 cells express both ERα and ERβ. Studies have reported various effects of E2 on Th17 cell differentiation. In mouse splenocytes, E2 inhibits Th17 cell differentiation and IL-17 production by inhibiting the expression of RORγt [22]. Similarly, in E2 deficiency-induced bone loss, the differentiation of Th17 cells was increased, accompanied by upregulation of STAT3, RORγt, and RORα and downregulation of FOXP3 [23]. However, studies performed by Andersson et al. on in experimental autoimmune arthritis (AA) have shown that estradiol treatment increases the amount of Th17 cells in lymph nodes during the early stage of arthritis development. In the advanced stage of the disease, estradiol acts in the opposite way, diminishing the number of Th17 cells in joints. The authors of the studies suggest the observed effect of estradiol action may be caused by the interference of E2 with CCR6-CCL20 (C-C chemokine receptor 6–C-C motif chemokine ligand 20) pathway, which is important for the migration of Th17 cells. E2 increased the expression of CCR6 on Th17 cells in lymph nodes as well as the expression of the corresponding CCL20 within lymph nodes [24]. Other studies on mice splenocytes demonstrated that ERα signaling increased IL-17A production in Th17 cells by upregulating the expression of IL-23R and promoting mitochondrial respiration and proliferation [25]. Deletion of ERα, but not ERβ, caused a significant decline in the production of IL-17A and surface expression of IL-23R on Th17 cells. These effects are realized through an increase in the relative expression of Let7f microRNA in Th17 cells. The findings of these studies show that ERα signaling regulates Th17 cell differentiation by influencing the Let7f/IL-23R pathway [26][27]. Thus, it seems that the influence of E2 on Th17 cells may depend on the environment in which the study is conducted and the type of disease analyzed, and the surrounding environment moderates the direction of E2′s influence on these cells.
It is known that the subset of Th17 cells is transient in nature. For example, in specific experimental conditions, CD4+ T cells may exhibit diminished IL-17 expression and upregulated IFN-γ expression [28]. In addition, Th17 lineage exhibits plasticity and can transdifferentiate into Treg cells [29]. FOXP3-expressing Treg cells, mostly represented by CD4+ T cells that express CD25 (IL-2 receptor α-chain), are important for controlling self-tolerance and immune homeostasis, but also suppress antitumor immune responses and favor tumor progression [30]. Th17 cells can act as a source of tumor-induced FOXP3+ cells, as it was shown by Downs-Canner et al. In addition to natural Treg and induced Treg cells formed from naïve precursors, suppressive IL-17A+ FOXP3+ and ex-Th17 FOXP3+ cells are converted from IL-17A+ FOXP3− cells in tumor-bearing mice [30]. Moreover, Th17/Th1 cells produce both IL-17 and IFNγ and Th17 cells stimulated by IL-12 can shift to Th17/Th1 [31][32]. Activated Th17 cells can produce IL-22 along with IL-17 [33], and represent a distinct population apart from Th22 cells [34]. Intestinal Th17 cells can differentiate into T follicular helper (Tfh) cells in Peyer’s patches [35]. The plasticity of Th17 cells has been described in detail elsewhere [36][37][38][39] (Figure 3).
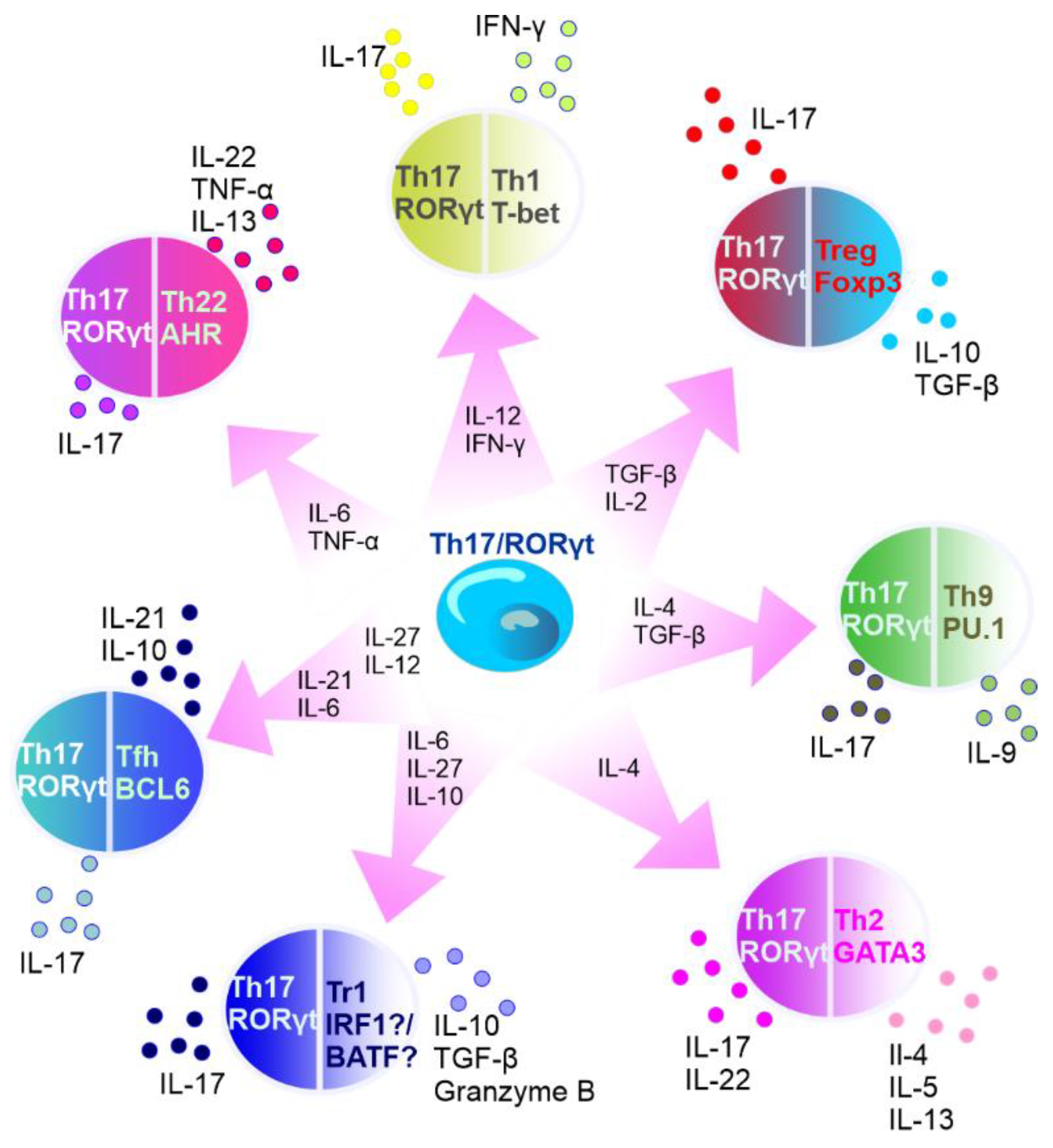
Figure 3. Summary of Th17 cell plasticity. In the circles, the names of T cells are presented with main transcription factors listed below the names. Outside the circles, the cytokines secreted by the specific cells are listed. Transcription factors: AhR—aryl hydrocarbon receptor, RORγt—retinoid-acid-receptor-related orphan nuclear receptor γ, IRF1—interferon regulatory factor 1, PU.1—transcription factor encoded by SPI1 gene, BATF—basic leucine zipper transcriptional factor ATF-like, BCL6—B-cell lymphoma 6, T-bet—member of T-box family of transcription factors, GATA3—GATA binding protein 3, and FOXP3—forkhead box P3. Cytokines: IL—interleukin, TNF-α—tumor necrosis factor α, IFN-γ—interferon γ, and TGF-β—transforming growth factor β. Cells: Tr1—type 1 regulatory T cells, Tfh—T follicular helper cells, Th—T helper cells, and Treg—T regulatory cells.
The phosphoinositide 3-kinase (PI3K)/serine/threonine-protein kinase (AKT) signaling pathway is involved in the processes of cell growth, differentiation, and apoptosis, and its activation is critical for the completion of cell cycle and cell differentiation. In addition, T cells proliferation and migration is also regulated by PI3K/AKT pathway. It has been shown that Th17 cell differentiation (both in vitro and in vivo) can be regulated by mTORC1 and mTORC2, mammalian targets of rapamycin (mTOR) complexes (via PI3K/AKT in different ways) [18][40]. Activation of PI3K and/or mTORC1 enhances the Th17 cell differentiation, but on the other hand, the inhibition of PI3K and/or mTORC1 in CD4+ T cells causes an increase in the differentiation of Treg cells [41].
Th17 cells can play both protective and pathogenic roles in immunity. The protective action of these effectors is related to the suppression of pathogens including Candida albicans and Staphylococcus aureus. However, it is believed that Th17 cells also induce inflammation and tissue damage [42].
2. Importance of Th17 Cells in Breast Cancer
A number of studies have confirmed the presence of Th17 cells in various types of cancers (e.g., breast, ovarian, colorectal, cervical cancer, and melanoma) and the significance of these cells in these diseases [43][44][45]. However, it is difficult to provide a clear description of the role played by Th17 cells in tumor development due to complex interactions occurring between cancer cells and the components of the host microenvironment [39]. Inflammation is often associated with cancer progression and actively contributes to the survival of cancer cells, angiogenesis, and metastasis [46]. It is known that tumor cells and cancer-associated fibroblasts (CAFs) create an inflammatory environment favorable for the recruitment of Th17 cells [47].
Although many studies have been carried out on Th17 cells, the role of these cells in breast cancer remains undefined [48]. Nevertheless, the majority of evidence indicates that Th17 cells exhibit prooncogenic properties in breast cancer. Clinical analyses have shown that the level of Th17 cells/IL-17 cytokine is usually altered in patients with breast cancer [43][49]. Compared to healthy donors, the level of Th17 cells is higher in the blood of breast cancer patients and correlates with elevated levels of C-X-C motif chemokine ligand (CXCL) 1. CXCL1, a proinflammatory chemokine produced by breast cancer cells, can promote cancer growth and development [50]. A positive correlation between the levels of IL-17 and macrophage infiltration inhibitory factor (MIF) has also been observed, and both IL-17 and MIF were linked with a high risk of developing breast cancer of aggressive molecular subtypes [51]. In breast tumors characterized by matrix metalloprotease (MMP) 11 expression by intratumoral mononuclear inflammatory cells, the level of expression of inflammatory factors, associated with distant metastasis development, such as IL-17 and NFκB (nuclear factor kappa-light-chain-enhancer of activated B cells), was shown to be significantly higher [52]. Infiltration by IL-17+ T cells in TNBC patients was associated with a poor recurrence-free survival [53]. Moreover, infiltration by Th17 cells is preferably observed in ER−, PR−, and TNBC tumors and is a poor prognostic factor [54]; it also correlates with failure of complete pathological response [55][56]. Another study demonstrated that an increased number of IL-17A-producing cells are found mainly in ER– and triple-negative/basal-like breast tumors [57]. On the other hand, high levels of ER suppress Th17 cell infiltration and IL-17 signal transduction, causing a reduction in PD-1/PD-L1 expression and CD8+ T cell infiltration in breast cancer [58]. In turn, a study by Horlock et al. showed that the number of circulating Th17 cells was the lowest in patients with HER2+ breast cancer compared to healthy controls and HER2– patients. An inverse relationship was also observed between the frequencies of Treg and Th17 cells in metastatic breast cancer with a significant reduction in the level of Treg cells during treatment with trastuzumab, whereas the level of Th17 cells was concomitantly increased [59]. On the other hand, a study investigated the distribution of IL-17-producing CD4+ T-cells in relation to Treg cells in tumor-infiltrating lymphocytes (TILs) and peripheral blood mononuclear cells (PBMCs) collected from breast cancer patients. The frequency of Th17 cells was found to be significantly higher in TILs than in PBMCs obtained from early breast cancer patients. In the TILs collected from advanced breast cancer patients, the frequency of Th17 cells was also significantly higher compared to that in PBMCs but lower compared to PBMCs from patients with early disease. Based on these findings, the authors concluded that the accumulation of Th17 and Treg cells in the tumor microenvironment of breast cancer occurred during the early stage of the disease. It was also indicated that Th17 cell infiltration gradually decreased but Treg cells continued to accumulate as the disease progressed [60]. However, studies on mice showed that the Th17 subpopulation was dominant in CD4+ T cells from TILs, and the population was also higher in the late tumor stages [61].
A meta-analysis of IL-17A estimation by immunohistochemistry, overall survival, and disease-free survival in patients with solid tumors indicated that in most of the cases these parameters were worse with higher levels of IL-17A. IL-17A was also associated with an advanced stage of cancer [48]. However, the association between the level of intratumoral Th17 cells and blood level of IL-17 was not clear. In addition, their effects did not seem to be unequivocal; thus, when Th17 cytokines (IL-17A and IL-17F), which were upregulated in TNBC, specifically in T cell noninflamed tumors, were exploited by the METABRIC transcriptomic dataset, a high expression of Th17 metagene was identified as an indicator of good prognosis of T cell noninflamed TNBC [62].
In addition to IL-17A, other IL-17 family members and their receptors have been analyzed in breast cancer patients. Mombelli et al. reported that mRNA expression of IL-17A and IL-17E receptor subunits was upregulated in breast cancers in comparison to normal samples. Furthermore, it seems that IL-17E, which is usually undetectable in normal breasts, is overexpressed in cancerous tissues [63]. It can also promote resistance to antimitotic and anti-EGFR therapies. In the breast cancer cell lines IJG-1731, BT20, and MDA-MB-468, EGFR phosphorylation is stimulated by epidermal growth factor and IL-17E. IL-17E also activates kinases that are crucial for EGFR signaling, such as PYK-2, Src, and STAT3 [64]. IL-17E and IL-17A induce cell proliferation and survival by activating pathways including c-RAF, ERK1/2, and p70S6 kinase, which also leads to docetaxel resistance [63]. Cochaud et al. authenticated that in human breast cancer cell lines recombinant IL-17A recruits the mitogen-activated protein kinases (MAPK) pathway by upregulating phosphorylated ERK1/2 which results in stimulation of cell proliferation, migration and invasion, and resistance to commonly used chemotherapeutic agents such as docetaxel [57].
A high level of IL-17B was found in patient biopsies, which was associated with a decrease in overall survival and with poor prognosis. Moreover, overexpression of IL-17RB was associated with reduced disease-free survival. Both overall and disease-free survival were reduced in patients with overexpression of IL-17B and IL-17RB [65]. In another study, the overexpression of IL-17RA and IL-17RB is associated with poor prognosis and shorter survival rate [66]. In breast cancer cell lines BT20, MDA-MB-468, and MCF-7, IL-17B induced resistance to paclitaxel, and activation of the extracellular signal-regulated kinase 1/2 (ERK1/2) pathway, leading to the upregulation of B-cell lymphoma 2 (Bcl-2) [65]. Moreover, IL-17RB and IL-17B amplification can promote tumorigenicity in breast cancer via the activation of NF-kB and Bcl-2. It has been shown that depletion of IL-17RB in tastuzumab-resistant cell lines ceased colony formation and retarded tumor growth in mice [66]. Recently, Bastid et al. comprehensively described the role of IL-17B and IL-17RB signaling pathways in cancer [67].
Kiyomi et al. reported that tumor tissues resected from breast cancer patients produced Th17 cytokines when cultured in three-dimensional gelatin polymer culture system [68]. Tumor cells and CAFs produce microenvironmental factors, such as RANTES (Regulated on Activation, Normal T-cell Expressed and Secreted) and monocyte chemoattractant protein-1 (MCP-1/CCL-2) chemoattractants, which mediate the recruitment of Th17 cells, and IL-23 and TGF-β, which are important factors of Th17 cell differentiation and generation. They also allow cell contact, inducing the generation and expansion of Th17 cells. Colony of Th17 cells in TILs obtained from patients produced high levels of IL-8, IL-17, and TNF and a low level of IL-6. Th17 clones expressed chemokine receptors such as CCR2, CCR4, CCR5, CCR6, CCR7, and CXC chemokine receptor (CXCR) 3, which are homeostatic chemokine receptors as well as trafficking receptors found commonly in other T cell lineages, including Treg cells [47]. Tumor production of IL-6 and TGF-β stimulated the differentiation of Th17 cells into CD25high/CD39/CD73 Th17 cells. Th17 CD25high cells accumulate in breast cancer tissue by recruitment via CCL20/CCR6. Intratumoral Th17 cells, which are also known as memory CD25high/CCR6+ Th17 cells, express IL-17, RORγ, FOXP3, CD39, and CD73. CD39 and CD73 are ectonucleotidases, which catalyze the transformation of ATP, and can lower T cell response. When these enzymes accumulate, they can weaken T cell immunity in breast cancer patients by suppressing CD4+ and CD8+ T cells, which worsens relapse-free and overall survival [69]. In blood samples and invasive ductal carcinoma (IDC) tissue collected from breast cancer patients, Th17-related molecules (IL-17A, RORC, and CCR6), produced by tumor-infiltrating CD4+ and CD8+ T lymphocytes, were observed to be upregulated. Angiogenic factors CXCL8, MMP-2, and MMP-9 and vascular endothelial growth factor (VEGF)-A were detected within the tumor and shown to be induced by IL-17, which correlated with poor prognosis. The accumulation of Treg and Th17 cells within an invasive breast tumor may promote the growth and survival of the tumor cells, and the presence of Treg cells and high levels of TGF-β may also favor the development of Th17 cells [70].
The genetic factors involved in the regulation of Th17 cell differentiation are currently being investigated. Numerous long noncoding RNAs (lncRNAs) are reported to regulate immune response in breast cancer patients [71]. One epigenetically dysregulated lncRNA (LINC01983) and four lncRNA regulators (UCA1, RP11-221J22.2, RP11-221J22.1, and RP1-212P9.3) were identified to act as prognostic biomarkers of luminal breast cancer by controlling the TNF signaling pathway, Th17 cell differentiation, and T cell migration [72].
Single-nucleotide polymorphisms (SNPs) of the IL-17 gene have been shown to be correlated with susceptibility to cancer [73]. Wang et al. analyzed SNPs of IL-17A and F genes and reported that rs2275913 polymorphism of IL-17A gene was associated with an increased risk of breast cancer in Chinese women [74]. However, Naeimi et al. indicated that polymorphisms of IL-17A and IL-17F genes have no significance in the susceptibility of women from southern Iran to breast cancer [75].
In addition, studies on animal models showed protumoral, prometastatic, and proangiogenic activity of IL-17 as well as the impact of this molecule on chemoresistance. A number of studies were conducted in the 4T1 mouse mammary gland tumor model, the growth of which is associated with high immune response including large leukocytosis, and lung and tumor infiltration by neutrophils [76]. Th17 lymphocytes were shown to be increased in the peripheral blood, spleen, and tumor tissue of 4T1 tumor-bearing mice [77][78]. PGE2 secreted by this tumor induced the production of IL-23 in the tumor microenvironment, leading to the expansion of Th17 cells [78]. In another study, the authors characterized T cells specific for 4T1 cancer and described them as receptor activator for nuclear factor κB ligand (RANKL)+ IL-17F+ CD4+ T cells [79]. Such cells arrive in the bone marrow before metastatic cells and build a premetastatic niche, which in effect leads to premetastatic osteolytic disease and bone metastases. 4T1-conditioned media support the differentiation of DCs to mature and activated multinucleated giant cells expressing TRAP and IL-23. These cytokines are involved in the activation of 4T1 tumor-specific T cells determined by RANKL and IL-17 production [80]. Moreover, the production of IL-17F and RANKL was only observed in cells derived from mice bearing 4T1 metastatic tumors, and not in cells from mice bearing 67NR nonmetastatic cells [80]. Administration of IL-17 in 4T1 tumor-bearing mice resulted in an increase in tumor size and a higher microvascular density [77]. Furthermore, a decrease in the levels of IL-17A caused by treatment with endothelin-1 receptor dual antagonist led to the slowdown of the growth of 4T1 tumor. In immunocompetent mice implanted with 4T1 cells, such treatment resulted in a reduced tumor growth and a decrease in the concentrations of proinflammatory TNF-α and IL-17 cytokines [81]. Similarly, knockdown of IL-17R in 4T1 mouse mammary gland cancer cells caused a reduction in tumor size and enhanced apoptosis [82]. Inhibition of IL-17 significantly reduced the metastases of spontaneously developing mammary gland carcinoma in MMTV-PyV MT mice with induced AA. In these mice, AA as well as lung and bone metastasis correlated with a high level of IL-17 [83]. In MCF-7, MDA-MB-157, MDA-MB-361, and MDA-MB-468 human breast cancer cell lines, high levels of IL-17RB as well as high IL-17RB mRNA expression have been observed. Depletion of IL-17RB resulted in inhibited colony formation and retarded MDA-MB-361 tumor growth in mice [66]. Inhibition of IL-17 also reduced the proliferation and colony formation as well as tumor growth, as revealed by chorioallantoic membrane assay (CAM) using MCF-7 cells [84]. At the same time, when stimulated with Th17 cells, MCF-7, MDA-MB-435, T47D, and MDA-MB 231 cells showed increased matrigel invasion [85].
In a murine study on mice bearing parental Cl66 murine mammary tumors and Cl66 cells resistant to doxorubicin (Cl66-Dox) or paclitaxel (Cl66-Pac) Wu et al. revealed the role of IL-17, CXCR2 ligands, and cancer-associated neutrophils in chemotherapy resistance and metastasis of breast cancer [86]. In tumor tissue of resistant models increased levels of IL-17R, CXCR2 chemokines, and CXCR2 were observed in comparison to C166 tumor tissue. What speaks for the significance of Th17 cells in chemoresistant cancer cells is the higher infiltration grade by Th17 and neutrophils in C166-Dox and C166-Pac models.
In addition, CD8+ T cells (splenocytes) from 4T1 tumor-bearing mice expressed IL-17, which promoted cell survival and reduced apoptosis. Addition of TGF-β and IL-6 caused a threefold higher IL-17 expression in CD8+ T cells from tumor-bearing mice than from naïve mice. A significant decrease in tumor size was also noted after blocking TGF-β and after depletion of CD8+ T lymphocytes. A similar reduction effect was observed on lung metastases [82].
It was shown that IL-17-producing γδ T cells and neutrophils synergistically promoted breast cancer metastasis in the mouse model of spontaneous metastasis [87]. IL-17+ γδT cells played an important role in oxidative metabolism, with increased mitochondrial mass and activity. Protumoral IL-17+ γδT cells selectively showed high lipid uptake and intracellular lipid storage and expanded in the tumors of obese mice [88].
IL-17E was also found to exhibit antitumor effects in mice lacking various T lymphocytes-bearing tumors, including breast cancer, but not in mice lacking both T and B lymphocytes. Treatment with IL-17E resulted in a significant increase in IL-5 serum levels and increased numbers of eosinophils in peripheral blood of tumor bearing mice. Also a significant increase in eosinophils was observed in spleens isolated from IL-17E-treated mice, which correlated with the antitumor activity of IL-17E in a dose-dependent manner. Moreover, B cells play also an important role in IL-17E-mediated antitumor activity. IL-17E activated signaling pathways in B cells in vitro [89]. In addition, breast cancer cells treated with IL-17E obtained from nonmalignant mammary epithelial cells-conditioned medium showed decreased colony formation [90]. Myeloid-derived suppressor cells (MDSCs), which are found at increased levels in breast cancer patients, were purified from mice bearing MCF-7 tumors and treated with IL-17. This treatment significantly induced the differentiation of MDSCs, inhibited their proliferation, and triggered apoptosis as well as inhibited the activation of STAT3 in these cells (Ma, Huang, and Kong, 2018).
The pro- and anti-cancer effect of Th17 cells and their cytokines has been reviewed by Fabre et al. [91] and Qianmeni et al. [92] and is summarized in Figure 4.
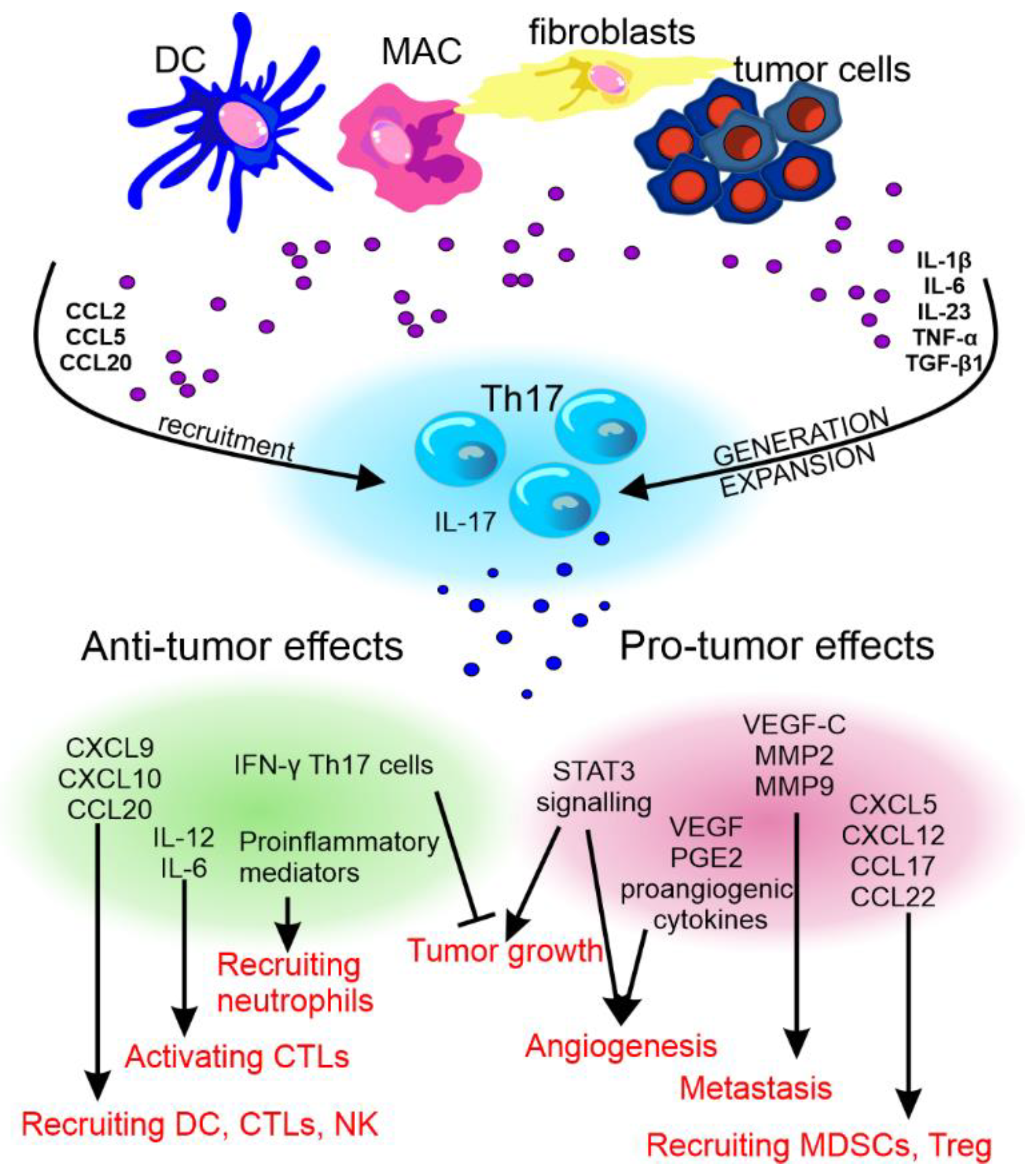
Figure 4. The anti- and pro-tumor effects of Th17 cells. The recruitment and differentiation of Th17 cells in the tumor environment is influenced by factors produced by dendritic cells (DCs), macrophages (MAC), fibroblasts, and cancer cells. Th17 cells differentiated in this way may show various effects on tumor development. CCL—C-C motif chemokine ligand, TNF-α—tumor necrosis factor α, TGF-β—transforming growth factor β, CXCL—C-X-C motif chemokine ligand, MMP—matrix metalloproteinase, STAT3—signal transducer and activator of transcription 3, VEGF—vascular endothelial growth factor, PGE2—prostaglandin E2, IFN-γ—interferon γ, IL—interleukin, CTLs—cytotoxic T lymphocytes, and NK—natural killer cells.
This entry is adopted from: 10.3390/cancers14153649
References
- Knochelmann, H.M.; Dwyer, C.; Bailey, S.; Amaya, S.M.; Elston, D.M.; Mazza-McCrann, J.M.; Paulos, C.M. When worlds collide: Th17 and Treg cells in cancer and autoimmunity. Cell. Mol. Immunol. 2018, 15, 458–469.
- Zhou, L.; Lopes, J.E.; Chong, M.; Ivanov, I.I.; Min, R.; Victora, G.; Shen, Y.; Du, J.; Rubtsov, Y.P.; Rudensky, A.Y.; et al. TGF-β-induced Foxp3 inhibits TH17 cell differentiation by antagonizing RORγt function. Nature 2008, 453, 236–240.
- Yosef, N.; Shalek, A.K.; Gaublomme, J.T.; Jin, H.; Lee, Y.; Awasthi, A.; Wu, C.; Karwacz, K.; Xiao, S.; Jorgolli, M.; et al. Dynamic regulatory network controlling TH17 cell differentiation. Nature 2013, 496, 461–468.
- Yang, X.O.; Panopoulos, A.D.; Nurieva, R.; Chang, S.H.; Wang, D.; Watowich, S.S.; Dong, C. STAT3 Regulates Cytokine-mediated Generation of Inflammatory Helper T Cells. J. Biol. Chem. 2007, 282, 9358–9363.
- Yang, X.O.; Pappu, B.P.; Nurieva, R.; Akimzhanov, A.; Kang, H.S.; Chung, Y.; Ma, L.; Shah, B.; Panopoulos, A.D.; Schluns, K.S.; et al. T Helper 17 Lineage Differentiation Is Programmed by Orphan Nuclear Receptors RORα and RORγ. Immunity 2008, 28, 29–39.
- Nurieva, R.; Yang, X.O.; Martinez, G.; Zhang, Y.; Panopoulos, A.; Ma, L.; Schluns, K.; Tian, Q.; Watowich, S.S.; Jetten, A.; et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature 2007, 448, 480–483.
- Chung, Y.; Chang, S.H.; Martinez, G.J.; Yang, X.O.; Nurieva, R.; Kang, H.S.; Ma, L.; Watowich, S.S.; Jetten, A.M.; Tian, Q.; et al. Critical Regulation of Early Th17 Cell Differentiation by Interleukin-1 Signaling. Immunity 2009, 30, 576–587.
- Chalmin, F.; Mignot, G.; Bruchard, M.; Chevriaux, A.; Végran, F.; Hichami, A.; Ladoire, S.; Derangère, V.; Vincent, J.; Masson, D.; et al. Stat3 and Gfi-1 Transcription Factors Control Th17 Cell Immunosuppressive Activity via the Regulation of Ectonucleotidase Expression. Immunity 2012, 36, 362–373.
- Nowatzky, J.; Manches, O.; Khan, S.A.; Godefroy, E.; Bhardwaj, N. Modulation of human Th17 cell responses through complement receptor 3 (CD11 b/CD18) ligation on monocyte-derived dendritic cells. J. Autoimmun. 2018, 92, 57–66.
- Segura, E.; Touzot, M.; Bohineust, A.; Cappuccio, A.; Chiocchia, G.; Hosmalin, A.; Dalod, M.; Soumelis, V.; Amigorena, S. Human Inflammatory Dendritic Cells Induce Th17 Cell Differentiation. Immunity 2013, 38, 336–348.
- Vroman, H.; van den Blink, B.; Kool, M. Mode of Dendritic Cell Activation: The Decisive Hand in Th2/Th17 Cell Differentiation. Implications in Asthma Severity? Immunobiology 2015, 220, 254–261.
- Schirmer, C.; Klein, C.; von Bergen, M.; Simon, J.C.; Saalbach, A. Human fibroblasts support the expansion of IL-17–producing T cells via up-regulation of IL-23 production by dendritic cells. Blood 2010, 116, 1715–1725.
- Pastor-Fernández, G.; Mariblanca, I.R.; Navarro, M.N. Decoding IL-23 Signaling Cascade for New Therapeutic Opportunities. Cells 2020, 9, 2044.
- Park, S.-H.; Jung, H.-J.; Kim, T.S. IL-33 changes CD25hi Tregs to Th17 cells through a dendritic cell-mediated pathway. Immunol. Lett. 2019, 218, 5–10.
- Shan, M.; Yuan, X.; Song, L.-Z.; Roberts, L.; Zarinkamar, N.; Seryshev, A.; Zhang, Y.; Hilsenbeck, S.; Chang, S.-H.; Dong, C.; et al. Cigarette Smoke Induction of Osteopontin (SPP1) Mediates T H 17 Inflammation in Human and Experimental Emphysema. Sci. Transl. Med. 2012, 4, 117ra9.
- Murugaiyan, G.; Mittal, A.; Weiner, H.L. Increased Osteopontin Expression in Dendritic Cells Amplifies IL-17 Production by CD4+ T Cells in Experimental Autoimmune Encephalomyelitis and in Multiple Sclerosis. J. Immunol. 2008, 181, 7480–7488.
- Zheng, Y.; Wang, Z.; Deng, L.; Yuan, X.; Ma, Y.; Zhang, G.; Gantier, M.P.; Liu, J.-P.; Shen, L.; Xu, D. Osteopontin promotes inflammation in patients with acute coronary syndrome through its activity on IL-17 producing cells. Eur. J. Immunol. 2012, 42, 2803–2814.
- Zhao, Q.; Cheng, W.; Xi, Y.; Cao, Z.; Xu, Y.; Wu, T.; Li, C.; Niu, X.; Chen, G. IFN-β regulates Th17 differentiation partly through the inhibition of osteopontin in experimental autoimmune encephalomyelitis. Mol. Immunol. 2017, 93, 20–30.
- Icer, M.A.; Gezmen-Karadag, M. The multiple functions and mechanisms of osteopontin. Clin. Biochem. 2018, 59, 17–24.
- Baaten, B.J.; Li, C.-R.; Bradley, L.M. Multifaceted regulation of T cells by CD44. Commun. Integr. Biol. 2010, 3, 508–512.
- Chen, G.; Zhang, X.; Li, R.; Fang, L.; Niu, X.; Zheng, Y.; He, D.; Xu, R.; Zhang, J.Z. Role of osteopontin in synovial Th17 differentiation in rheumatoid arthritis. Arthritis Care Res. 2010, 62, 2900–2908.
- Chen, R.-Y.; Fan, Y.-M.; Zhang, Q.; Liu, S.; Li, Q.; Ke, G.-L.; Li, C.; You, Z. Estradiol Inhibits Th17 Cell Differentiation through Inhibition of RORγT Transcription by Recruiting the ERα/REA Complex to Estrogen Response Elements of the RORγT Promoter. J. Immunol. 2015, 194, 4019–4028.
- Tyagi, A.M.; Srivastava, K.; Mansoori, M.N.; Trivedi, R.; Chattopadhyay, N.; Singh, D. Estrogen Deficiency Induces the Differentiation of IL-17 Secreting Th17 Cells: A New Candidate in the Pathogenesis of Osteoporosis. PLoS ONE 2012, 7, e44552.
- Andersson, A.; Stubelius, A.; Karlsson, M.N.; Engdahl, C.; Erlandsson, M.; Grahnemo, L.; Lagerquist, M.K.; Islander, U. Estrogen regulates T helper 17 phenotype and localization in experimental autoimmune arthritis. Arthritis Res. Ther. 2015, 17, 32.
- Fuseini, H.; Cephus, J.-Y.; Wu, P.; Davis, J.B.; Contreras, D.C.; Gandhi, V.D.; Rathmell, J.C.; Newcomb, D.C. ERα Signaling Increased IL-17A Production in Th17 Cells by Upregulating IL-23R Expression, Mitochondrial Respiration, and Proliferation. Front. Immunol. 2019, 10, 2740.
- Fuseini, H.; Cephus, J.; Davis, J.B.; Peebles, S.; Newcomb, D.C. Estrogen receptor α signaling promotes IL-17A production in Th17 cells through the Let7/IL-23R signaling pathway. J. Allergy Clin. Immunol. 2019, 143, AB213.
- Newcomb, D.C.; Cephus, J.Y.; Boswell, M.G.; Fahrenholz, J.M.; Langley, E.W.; Feldman, A.S.; Zhou, W.; Dulek, D.E.; Goleniewska, K.; Woodward, K.B.; et al. Estrogen and progesterone decrease let-7f microRNA expression and increase IL-23/IL-23 receptor signaling and IL-17A production in patients with severe asthma. J. Allergy Clin. Immunol. 2015, 136, 1025–1034.e11.
- Kurschus, F.C.; Croxford, A.L.; Heinen, A.P.; Wörtge, S.; Ielo, D.; Waisman, A. Genetic proof for the transient nature of the Th17 phenotype. Eur. J. Immunol. 2010, 40, 3336–3346.
- Gagliani, N.; Amezcua Vesely, M.C.; Iseppon, A.; Brockmann, L.; Xu, H.; Palm, N.W.; De Zoete, M.R.; Licona-Limón, P.; Paiva, R.S.; Ching, T.; et al. Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature 2015, 523, 221–225.
- Downs-Canner, S.; Berkey, S.; Delgoffe, G.M.; Edwards, R.P.; Curiel, T.; Odunsi, K.; Bartlett, D.L.; Obermajer, N. Suppressive IL-17A+Foxp3+ and ex-Th17 IL-17AnegFoxp3+ Treg cells are a source of tumour-associated Treg cells. Nat. Commun. 2017, 8, 14649.
- Annunziato, F.; Cosmi, L.; Santarlasci, V.; Maggi, L.; Liotta, F.; Mazzinghi, B.; Parente, E.; Filì, L.; Ferri, S.; Frosali, F.; et al. Phenotypic and functional features of human Th17 cells. J. Exp. Med. 2007, 204, 1849–1861.
- Kotake, S.; Yago, T.; Kobashigawa, T.; Nanke, Y. The Plasticity of Th17 Cells in the Pathogenesis of Rheumatoid Arthritis. J. Clin. Med. 2017, 6, 67.
- Liang, S.C.; Tan, X.-Y.; Luxenberg, D.P.; Karim, R.; Dunussi-Joannopoulos, K.; Collins, M.; Fouser, L.A. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J. Exp. Med. 2006, 203, 2271–2279.
- Lindahl, H.; Olsson, T. Interleukin-22 Influences the Th1/Th17 Axis. Front. Immunol. 2021, 12, 618110.
- Hirota, K.; Turner, J.-E.; Villa, M.; Duarte, J.H.; Demengeot, J.; Steinmetz, O.M.; Stockinger, B. Plasticity of TH17 cells in Peyer’s patches is responsible for the induction of T cell–dependent IgA responses. Nat. Immunol. 2013, 14, 372–379.
- Stadhouders, R.; Lubberts, E.; Hendriks, R.W. A cellular and molecular view of T helper 17 cell plasticity in autoimmunity. J. Autoimmun. 2018, 87, 1–15.
- Muranski, P.; Restifo, N.P. Essentials of Th17 cell commitment and plasticity. Blood 2013, 121, 2402–2414.
- Agalioti, T.; Villablanca, E.J.; Huber, S.; Gagliani, N. TH17 cell Plasticity: The Role of Dendritic Cells and Molecular Mechanisms. J. Autoimmun. 2018, 87, 50–60.
- Cerboni, S.; Gehrmann, U.; Preite, S.; Mitra, S. Cytokine-regulated Th17 plasticity in human health and diseases. Immunology 2020, 163, 3–18.
- Nagai, S.; Kurebayashi, Y.; Koyasu, S. Role of PI3K/Akt and mTOR complexes in Th17 cell differentiation. Ann. N. Y. Acad. Sci. 2013, 1280, 30–34.
- Sauer, S.; Bruno, L.; Hertweck, A.; Finlay, D.; Leleu, M.; Spivakov, M.; Knight, Z.A.; Cobb, B.S.; Cantrell, D.; O’Connor, E.; et al. T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc. Natl. Acad. Sci. USA 2008, 105, 7797–7802.
- Céline, M.; Dubéa, A.L.; Brewstera, T.Z.B. T Helper Cell Polarization in Healthy People: Implications for Cardiovascular Disease. Bone 2012, 23, 1–7.
- Yang, L.; Qi, Y.; Hu, J.; Tang, L.; Zhao, S.; Shan, B. Expression of Th17 Cells in Breast Cancer Tissue and Its Association with Clinical Parameters. Cell Biophys. 2011, 62, 153–159.
- Kryczek, I.; Banerjee, M.; Cheng, P.; Vatan, L.; Szeliga, W.; Wei, S.; Huang, E.; Finlayson, E.; Simeone, D.; Welling, T.H.; et al. Phenotype, distribution, generation, and functional and clinical relevance of Th17 cells in the human tumor environments. Blood 2009, 114, 1141–1149.
- Wilke, C.M.; Kryczek, I.; Wei, S.; Zhao, E.; Wu, K.; Wang, G.; Zou, W. Th17 cells in cancer: Help or hindrance? Carcinogenesis 2011, 32, 643–649.
- DeNardo, D.G.; Johansson, M.; Coussens, L.M. Immune cells as mediators of solid tumor metastasis. Cancer Metastasis Rev. 2007, 27, 11–18.
- Su, X.; Ye, J.; Hsueh, E.C.; Zhang, Y.; Hoft, D.F.; Peng, G. Tumor Microenvironments Direct the Recruitment and Expansion of Human Th17 Cells. J. Immunol. 2009, 184, 1630–1641.
- Wang, S.; Li, Z.; Hu, G. Prognostic role of intratumoral IL-17A expression by immunohistochemistry in solid tumors: A meta-analysis. Oncotarget 2017, 8, 66382–66391.
- Allaoui, R.; Hagerling, C.; Desmond, E.; Warfvinge, C.-F.; Jirström, K.; Leandersson, K. Infiltration of Γδ T cells, IL-17+ T cells and FoxP3+ T cells in human breast cancer. Cancer Biomark. 2018, 20, 395–409.
- Ma, K.; Yang, L.; Shen, R.; Kong, B.; Chen, W.; Liang, J.; Tang, G.; Zhang, B. Th17 cells regulate the production of CXCL1 in breast cancer. Int. Immunopharmacol. 2018, 56, 320–329.
- Avalos-Navarro, G.; Muñoz-Valle, J.F.; Daneri-Navarro, A.; Quintero-Ramos, A.; Franco-Topete, R.A.; Morán-Mendoza, A.D.J.; Oceguera-Villanueva, A.; Bautista-Herrera, L.A.; Topete-Camacho, A.; Del Toro-Arreola, A. Circulating soluble levels of MIF in women with breast cancer in the molecular subtypes: Relationship with Th17 cytokine profile. Clin. Exp. Med. 2019, 19, 385–391.
- Eiró, N.; González, L.; González, L.O.; Fernandez-Garcia, B.; Lamelas, M.L.; Marín, L.; González-Reyes, S.; del Casar, J.M.; Vizoso, F.J. Relationship between the Inflammatory Molecular Profile of Breast Carcinomas and Distant Metastasis Development. PLoS ONE 2012, 7, e49047.
- Welsh, J. Function of the vitamin D endocrine system in mammary gland and breast cancer. Mol. Cell. Endocrinol. 2017, 453, 88–95.
- Chen, W.-C.; Lai, Y.-H.; Chen, H.-Y.; Guo, H.-R.; Su, I.-J.; Chen, H.H.W. Interleukin-17-producing cell infiltration in the breast cancer tumour microenvironment is a poor prognostic factor. Histopathology 2013, 63, 225–233.
- Kaur, R.P.; Vasudeva, K.; Singla, H.; Benipal, R.P.S.; Khetarpal, P.; Munshi, A. Analysis of pro- and anti-inflammatory cytokine gene variants and serum cytokine levels as prognostic markers in breast cancer. J. Cell. Physiol. 2018, 233, 9716–9723.
- Kaewkangsadan, V.; Verma, C.; Eremin, J.M.; Cowley, G.; Ilyas, M.; Eremin, O. Crucial Contributions by T Lymphocytes (Effector, Regulatory, and Checkpoint Inhibitor) and Cytokines (TH1, TH2, and TH17) to a Pathological Complete Response Induced by Neoadjuvant Chemotherapy in Women with Breast Cancer. J. Immunol. Res. 2016, 2016, 4757405.
- Cochaud, S.; Giustiniani, J.; Thomas, C.; Laprevotte, E.; Garbar, C.; Savoye, A.-M.; Curé, H.; Mascaux, C.; Alberici, G.; Bonnefoy, N.; et al. IL-17A is produced by breast cancer TILs and promotes chemoresistance and proliferation through ERK1/2. Sci. Rep. 2013, 3, 3456.
- Shuai, C.; Yang, X.; Pan, H.; Han, W. Estrogen Receptor Downregulates Expression of PD-1/PD-L1 and Infiltration of CD8+ T Cells by Inhibiting IL-17 Signaling Transduction in Breast Cancer. Front. Oncol. 2020, 10, 582863.
- Horlock, C.; Stott, B.; Dyson, P.J.; Morishita, M.; Coombes, R.C.; Savage, P.; Stebbing, J. The effects of trastuzumab on the CD4+CD25+FoxP3+ and CD4+IL17A+ T-cell axis in patients with breast cancer. Br. J. Cancer 2009, 100, 1061–1067.
- Wang, J.; Cai, D.; Ma, B.; Wu, G.; Wu, J. Skewing the Balance of Regulatory T-Cells and T-Helper 17 Cells in Breast Cancer Patients. J. Int. Med. Res. 2011, 39, 691–701.
- Huang, Y.; Ma, C.; Zhang, Q.; Ye, J.; Wang, F.; Zhang, Y.; Hunborg, P.; Varvares, M.A.; Hoft, D.F.; Hsueh, E.C.; et al. CD4+ and CD8+ T cells have opposing roles in breast cancer progression and outcome. Oncotarget 2015, 6, 17462–17478.
- Faucheux, L.; Grandclaudon, M.; Perrot-Dockès, M.; Sirven, P.; Berger, F.; Hamy, A.; Fourchotte, V.; Vincent-Salomon, A.; Mechta-Grigoriou, F.; Reyal, F.; et al. A multivariate Th17 metagene for prognostic stratification in T cell non-inflamed triple negative breast cancer. OncoImmunology 2019, 8, e1624130.
- Mombelli, S.; Cochaud, S.; Merrouche, Y.; Garbar, C.; Antonicelli, F.; Laprevotte, E.; Alberici, G.; Bonnefoy, N.; Eliaou, J.-F.; Bastid, J.; et al. IL-17A and its homologs IL-25/IL-17E recruit the c-RAF/S6 kinase pathway and the generation of pro-oncogenic LMW-E in breast cancer cells. Sci. Rep. 2015, 5, 11874.
- Merrouche, Y.; Fabre, J.; Cure, H.; Garbar, C.; Fuselier, C.; Bastid, J.; Antonicelli, F.; Al-Daccak, R.; Bensussan, A.; Giustiniani, J. IL-17E synergizes with EGF and confers in vitro resistance to EGFR-targeted therapies in TNBC cells. Oncotarget 2016, 7, 53350–53361.
- Laprevotte, E.; Cochaud, S.; du Manoir, S.; Lapierre, M.; Dejou, C.; Philippe, M.; Giustiniani, J.; Frewer, K.A.; Sanders, A.J.; Jiang, W.G.; et al. The IL-17B-IL-17 receptor B pathway promotes resistance to paclitaxel in breast tumors through activation of the ERK1/2 pathway. Oncotarget 2017, 8, 113360–113372.
- Huang, C.-K.; Yang, C.-Y.; Jeng, Y.-M.; Chen, C.-L.; Wu, H.-H.; Chang, Y.-C.; Ma, C.; Kuo, W.-H.; Chang, K.-J.; Shew, J.-Y.; et al. Autocrine/paracrine mechanism of interleukin-17B receptor promotes breast tumorigenesis through NF-κB-mediated antiapoptotic pathway. Oncogene 2013, 33, 2968–2977.
- Bastid, J.; Dejou, C.; Docquier, A.; Bonnefoy, N. The Emerging Role of the IL-17B/IL-17RB Pathway in Cancer. Front. Immunol. 2020, 11, 718.
- Kiyomi, A.; Makita, M.; Ozeki, T.; Li, N.; Satomura, A.; Tanaka, S.; Onda, K.; Sugiyama, K.; Iwase, T.; Hirano, T. Characterization and Clinical Implication of Th1/Th2/Th17 Cytokines Produced from Three-Dimensionally Cultured Tumor Tissues Resected from Breast Cancer Patients. Transl. Oncol. 2015, 8, 318–326.
- Thibaudin, M.; Chaix, M.; Boidot, R.; Végran, F.; Derangère, V.; Limagne, E.; Berger, H.; Ladoire, S.; Apetoh, L.; Ghiringhelli, F. Human ectonucleotidase-expressing CD25highTh17 cells accumulate in breast cancer tumors and exert immunosuppressive functions. OncoImmunology 2015, 5, e1055444.
- Benevides, L.; Cardoso, C.R.B.; Tiezzi, D.G.; Marana, H.R.C.; Andrade, J.M.; Silva, J.S. Enrichment of regulatory T cells in invasive breast tumor correlates with the upregulation of IL-17A expression and invasiveness of the tumor. Eur. J. Immunol. 2013, 43, 1518–1528.
- Mathias, C.; Muzzi, J.C.D.; Antunes, B.B.; Gradia, D.F.; Castro, M.A.A.; de Oliveira, J.C. Unraveling Immune-Related lncRNAs in Breast Cancer Molecular Subtypes. Front. Oncol. 2021, 11, 1936.
- Zhao, H.; Liu, X.; Yu, L.; Lin, S.; Zhang, C.; Xu, H.; Leng, Z.; Huang, W.; Lei, J.; Li, T.; et al. Comprehensive landscape of epigenetic-dysregulated lncRNAs reveals a profound role of enhancers in carcinogenesis in BC subtypes. Mol. Ther.-Nucleic Acids 2020, 23, 667–681.
- Dai, Z.-M.; Zhang, T.-S.; Lin, S.; Zhang, W.-G.; Liu, J.; Cao, X.-M.; Li, H.-B.; Wang, M.; Liu, X.-H.; Liu, K.; et al. Role of IL-17A rs2275913 and IL-17F rs763780 polymorphisms in risk of cancer development: An updated meta-analysis. Sci. Rep. 2016, 6, 20439.
- Wang, L.; Jiang, Y.; Zhang, Y.; Wang, Y.; Huang, S.; Wang, Z.; Tian, B.; Yang, Y.; Jiang, W.; Pang, D. Association Analysis of IL-17A and IL-17F Polymorphisms in Chinese Han Women with Breast Cancer. PLoS ONE 2012, 7, e34400.
- Naeimi, S.; Erfani, N.; Ardekani, A.M.; Ghaderi, A. Variation of IL-17A and IL-17F Genes in Patients with Breast Cancer in a Population from Southern Iran. Adv. Environ. Biol. 2014, 8, 892–897.
- Dupre, S.A.; Hunter, K.W. Murine mammary carcinoma 4T1 induces a leukemoid reaction with splenomegaly: Association with tumor-derived growth factors. Exp. Mol. Pathol. 2007, 82, 12–24.
- Du, J.-W.; Xu, K.-Y.; Fang, L.-Y.; Qi, X.-L. Interleukin-17, produced by lymphocytes, promotes tumor growth and angiogenesis in a mouse model of breast cancer. Mol. Med. Rep. 2012, 6, 1099–1102.
- Qian, X.; Gu, L.; Ning, H.; Zhang, Y.; Hsueh, E.C.; Fu, M.; Hu, X.; Wei, L.; Hoft, D.F.; Liu, J. Increased Th17 Cells in the Tumor Microenvironment Is Mediated by IL-23 via Tumor-Secreted Prostaglandin E2. J. Immunol. 2013, 190, 5894–5902.
- Monteiro, A.C.; Bonomo, A. Dendritic cells development into osteoclast-type APCs by 4T1 breast tumor T cells milieu boost bone consumption. Bone 2020, 143, 115755.
- Monteiro, A.C.; Leal, A.C.; Gonçalves-Silva, T.; Mercadante, A.C.T.; Kestelman, F.; Chaves, S.; Azevedo, R.B.; Monteiro, J.P.; Bonomo, A. T Cells Induce Pre-Metastatic Osteolytic Disease and Help Bone Metastases Establishment in a Mouse Model of Metastatic Breast Cancer. PLoS ONE 2013, 8, e68171.
- Jewell, A.N.; Swamydas, M.; Castillo, C.I.; Wyan, H.; Allen, L.D.; McDermott, K.A.; Eddy, J.M.; Dréau, D. The Endothelin Axis Stimulates the Expression of Pro-Inflammatory Cytokines and Pro-Migratory Molecules in Breast Cancer. Cancer Investig. 2010, 28, 932–943.
- Nam, J.-S.; Terabe, M.; Kang, M.-J.; Chae, H.; Voong, N.; Yang, Y.-A.; Laurence, A.; Michalowska, A.; Mamura, M.; Lonning, S.; et al. Transforming Growth Factor β Subverts the Immune System into Directly Promoting Tumor Growth through Interleukin-17. Cancer Res. 2008, 68, 3915–3923.
- Das Roy, L.; Ghosh, S.; Pathangey, L.B.; Tinder, T.L.; Gruber, H.E.; Mukherjee, P. Collagen induced arthritis increases secondary metastasis in MMTV-PyV MT mouse model of mammary cancer. BMC Cancer 2011, 11, 365.
- Kim, G.; Khanal, P.; Lim, S.-C.; Yun, H.J.; Ahn, S.-G.; Ki, S.H.; Choi, H.S. Interleukin-17 induces AP-1 activity and cellular transformation via upregulation of tumor progression locus 2 activity. Carcinogenesis 2012, 34, 341–350.
- Zhu, X.; Mulcahy, L.A.; Mohammed, R.A.A.; Lee, A.H.S.; Franks, H.A.; Kilpatrick, L.; Yilmazer, A.; Paish, E.C.; Ellis, I.O.; Patel, P.M.; et al. IL-17 expression by breast-cancer-associated macrophages: IL-17 promotes invasiveness of breast cancer cell lines. Breast Cancer Res. 2008, 10, R95.
- Wu, L.; Awaji, M.; Saxena, S.; Varney, M.L.; Sharma, B.; Singh, R.K. IL-17–CXC Chemokine Receptor 2 Axis Facilitates Breast Cancer Progression by Up-Regulating Neutrophil Recruitment. Am. J. Pathol. 2019, 190, 222–233.
- Coffelt, S.B.; Kersten, K.; Doornebal, C.W.; Weiden, J.; Vrijland, K.; Hau, C.-S.; Verstegen, N.J.M.; Ciampricotti, M.; Hawinkels, L.J.A.C.; Jonkers, J.; et al. IL-17-producing γδ T cells and neutrophils conspire to promote breast cancer metastasis. Nature 2015, 522, 345–348.
- Lopes, N.; McIntyre, C.; Martin, S.; Raverdeau, M.; Sumaria, N.; Kohlgruber, A.C.; Fiala, G.J.; Agudelo, L.Z.; Dyck, L.; Kane, H.; et al. Distinct metabolic programs established in the thymus control effector functions of γδ T cell subsets in tumor microenvironments. Nat. Immunol. 2021, 22, 179–192.
- Benatar, T.; Cao, M.Y.; Lee, Y.; Lightfoot, J.; Feng, N.; Gu, X.; Lee, V.; Jin, H.; Wang, M.; Wright, J.A.; et al. IL-17E, a proinflammatory cytokine, has antitumor efficacy against several tumor types in vivo. Cancer Immunol. Immunother. 2009, 59, 805–817.
- Furuta, S.; Jeng, Y.-M.; Zhou, L.; Huang, L.; Kuhn, I.; Bissell, M.J.; Lee, W.-H. IL-25 Causes Apoptosis of IL-25R–Expressing Breast Cancer Cells without Toxicity to Nonmalignant Cells. Sci. Transl. Med. 2011, 3, 78ra31.
- Fabre, J.A.S.; Giustiniani, J.; Garbar, C.; Merrouche, Y.; Antonicelli, F.; Bensussan, A. The Interleukin-17 Family of Cytokines in Breast Cancer. Int. J. Mol. Sci. 2018, 19, 3880.
- Qianmei, Y.; Zehong, S.; Guang, W.; Hui, L.; Lian, G. Recent advances in the role of Th17/Treg cells in tumor immunity and tumor therapy. Immunol. Res. 2021, 69, 398–414.
More