Tautomerism is typically this entry e relocation of a light atom, often hydrogen, at the same time as a rearrangement of the electronic structure. Typical examples are acetylacetone and acetone. This entry gives a short introduction to how to detect and describe a tautomeric equilibrium using Nuclear Magnetic Resonance (NMR). Typical parameters to be used are chemical shifts (1H,13C,15N,19F and 17O) one -bond coupling constants and isotope effects on chemical shifts.
- Tautomeric equilibria
- NMR
- chemical shifts
- coupling constants
- isotope effects
Introduction
Tautomerism is typically the relocation of a light atom, often hydrogen, at the same time as a rearrangement of the electronic structure. Typical examples are acetylacetone and acetone. In the latter molecule a keto-enol equilibrium is present (Figure 1), but the enol form is in a very low amount and not “visible”.
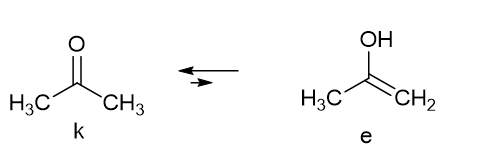
Figure 1. Acetone tautomeric equilibrium.
In the case of acetylacetone (as a representative of the broader group of β-diketones) two equilibria takes place, a keto-enol one, which is slow at the NMR time scale (which means that separate resonances from the two tautomers can be observed) and a fast movement of the enol proton from one oxygen to the other
(Figure 2).
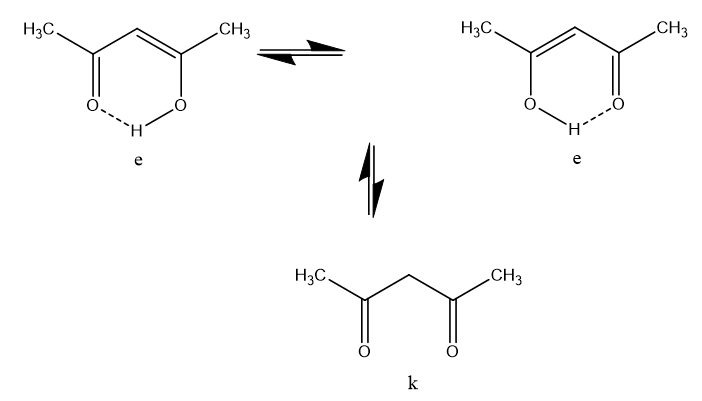
Figure 2. Tautomeric species of acetylacetone.
This latter equilibrium is very fast (the barrier is very low). It is not visible as such, but can be inferred from the fact that the two methyl resonances have the same chemical shift (see Figure 3). However, this is not the case in unsymmetrical cases. It is already clear that it can be difficult to detect the presence of an equilibrium. Could other spectroscopic techniques be useful? Yes, Infra Red spectroscopy is usually fast enough to detect the two tautomers and so is UV-VIS spectroscopy. However, Infra Red spectra of tautomeric compounds with two or more forms become very complex to interpret and for UV-VIS both forms may not have suitable chromophores. NMR clearly suffers from being a “slow” technique and will typically only provide average chemical shifts and coupling constants.
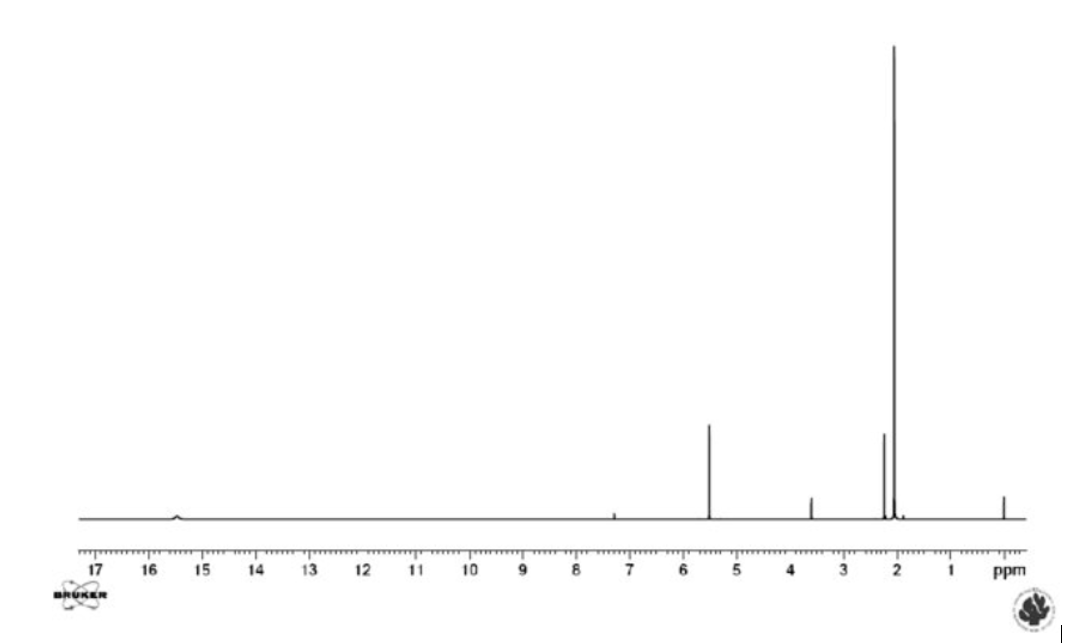
Figure 3. 1H NMR spectrum of acetylacetone in CDCl3. The resonances belonging to the keto form are those at 3.6 and 2.2 ppm.
Using NMR we are concentrating on chemical shifts, coupling constants and isotope effects on chemical shifts and temperature can be of additional help. Furthermore, as we often only observe an average signal, it is very useful to know the parameters of both tautomers. Density Functional Theory (DFT) calculations may be of great help. It is important that tautomeric equilibria can occur both in solution and in the solid state. [1]
Chemical shifts primarily used in tautomeric studies are 1H, 13C, 15N and 19F (if present) and to a much lesser degree 17O. Common for the latter four nuclei is a very large chemical shift range.
1H and 13C chemical shifts
1H is especially interesting when looking at XH protons (X=O, N or S) and when a large chemical shift difference is present between the two tautomers. A case is β-thioxoketones in which a large chemical shift difference is found between OH and SH chemical shifts, as well as large chemical shift differences between 13C chemical shifts of C=S and CSH exist (see Table 1). Furthermore, large equilibrium isotope effects are also seen in this type of tautomeric equilibrium (see later). However, one may easily make mistakes as in the present case by only assuming the traditional two types of tautomers (A and B of Figure 4). It turns out that also an open form with the SH not hydrogen bonded does exist (form C of Figure 4). [2]
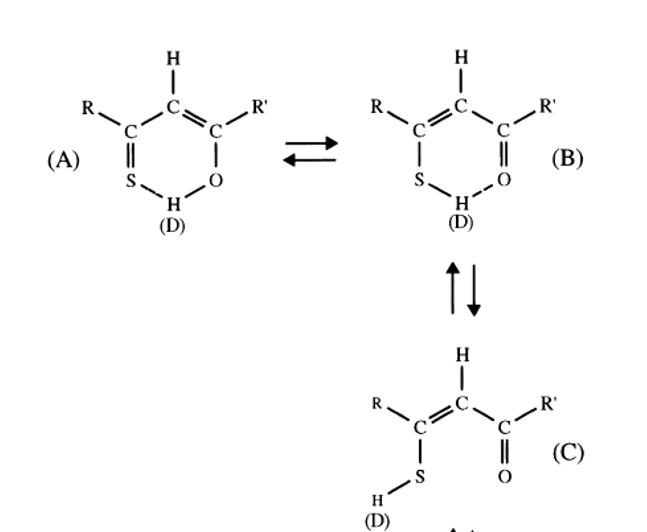
Figure 4. β-thixoketones. With R and R` equal to CH3 it is thioacetylacetone. D is referring to deuteriation (see isotope effects).
Table 1. Chemical shifts of thioacetylacetone from Ref. 2.
|
|
δCS |
δCSH |
δCO |
δCOH |
δOH |
δSH |
|
A |
217 |
|
|
186 |
15.2 |
|
|
B |
|
163 |
198 |
|
|
6a |
|
C |
|
158 |
193 |
|
|
3.32 |
- Estimated by comparison with model compounds
It is of course important to know the chemical shifts of the two tautomers. Chemical shifts for one of the two can normally be obtained from solid state NMR spectra. [3] Model compounds have also been suggested, but in that case the monitoring atom has to be far from the site of tautomeric action. [4] The chemicals shifts of both tautomeric species may be estimated from theoretical calculations (see later).
19F chemical shifts
The very large chemical shift range of 19F makes it well suited for detecting of tautomeric equilibria. A good example is fluorosubstituted o-hydroxyazo compounds (see Figure 5). [4]
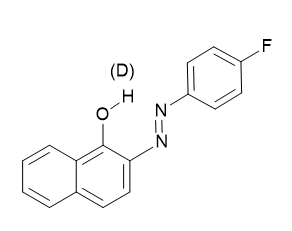
Figure 5. Only one tautomer is shown. For deuterium isotope effects on 19F chemical shifts see later.
Other examples are fluorosubstituted fuchsones and benzaurins [5] or fluoro-X-benzofurans. [6]
Also 13C-19F coupling constants may be used in the characterization of tautomeric systems.[7] This is building on the fact the one-bond C-F coupling depend strongly on the double character of the C-F bond.
17O chemical shifts
The 17O resonances suffer from broad lines, but this is normally compensated by the very large chemical shift range. [8] Examples of b-diketones and 1,8-dihydroxy2-acetyl, 3,6-dimethylnaphthalene are given in Ref. [9]
Coupling constants
A much used parameter is the 1J(N,H) in nitrogen containing compounds with an NH motif. This type of coupling is typically around 90 Hz. [10] 1J(N,H) can be used in all compounds containing an NH group taking part in tautomerism. A typical examples is “ o-hydroxy Schiff bases” see Figure 6. [10]
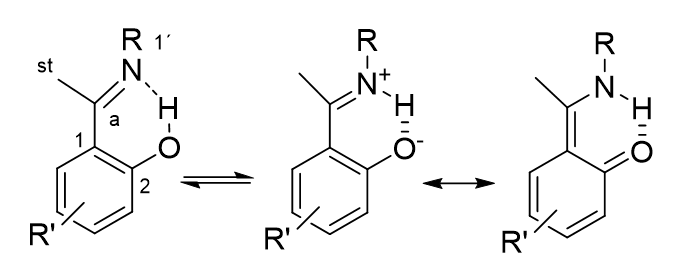
Figure 6. o-Hydroxy Schiff bases.
A typical problem to consider is NH exchange with the surrounding leading to too small coupling constants. Temperature studies may be helpful in eliminating this problem. For cases in which suitable model compounds are not available, 1J(N,H) can be predicted based on calculated NH bond lengths. [11]
Isotope effects on chemical shifts
Two types of isotope effects on chemical shifts, primary and secondary can be applied. They are defined as follows in case of deuterium as the isotope : primary , PΔH(D) = δH-δD and secondary, nΔX(D) = δX(H)-δX(D). n is the number of bonds between the deuterium and the atom in question, X. The most common isotope to be used is deuterium, as also indicated in the definition. In case of an equilibrium, the isotope effects are different from the so called intrinsic isotope effects, which are found in “static” cases.
Equilibrium isotope effects
The definition of the equilibrium isotope effects is expressed as:
ΔXH(D) x (δCM- δCPT) in which ΔXH(D is the change in the equilibrium upon deuteriation. The full equation was given early on by Bordner et al. [12] :
ΔC(D) = (δCA - δCB ) x (a - aD ) + [aD x ΔCA(D)i + (1- aD ) x ΔCB(D) i] Eq.1
The first term in this equation corresponds to the equilibrium isotope effect, whose magnitude depends on the change in the position of equilibrium (a - aD ) due to substitution of H by D and on the difference in the magnitude of the chemical shifts of the given carbon in the A and B forms. The second term represents the direct effect of the isotopic substitution on the chemical shifts (intrinsic isotope effect, ΔC(D)i ). The equilibrium term is a consequence of the potential energy diagram Figure 7. Deuteriation will in a double well potential lead to a change in the equilibrium. This means that the chemical shift difference comes into play multiplied with the change in the equilibrium upon deuteriation (see Eq. 1)
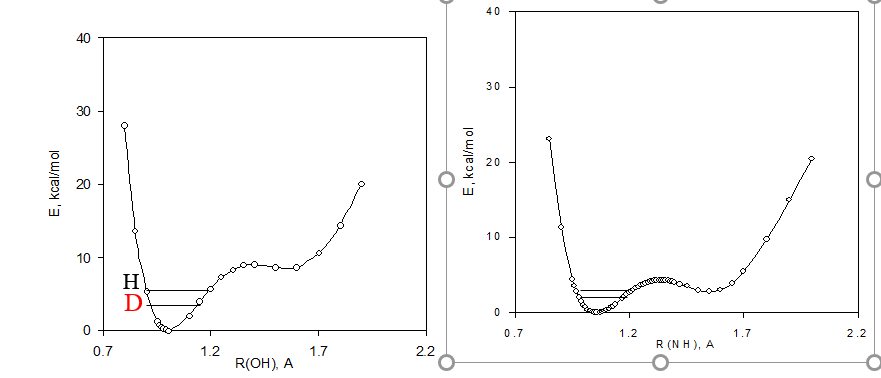
Figure 7. Potential energy diagrams including zero point energies for H and d species for a tautomeric equilibrium of o-hydroy Schiff bases.
The Limbach group has used an approach based on bond valences. See e.g. Ref. [13]
Reviews dealing with equilibrium isotope effects can be found. [11][14][15][16][17][18]
Primary deuterium isotope effects
The sign of the primary deuterium isotope effect can be used to estimate hydrogen bond strength. Positive primary isotope effects are for weak to medium strong isotope effects, whereas a negative isotope effects was claimed to be an indicator of a strong hydrogen bond. [19] However, if equilibrium is occurring positive as well as negative effects may occur. [11]
Secondary deuterium isotope effects
Deuterium isotope effects on 13C chemical shifts
Secondary deuterium isotope effects have a characteristic S-shaped dependence if plotted vs. the mole fraction as illustrated in Figure 8. [20] It is also obvious that at the maximum and minimum the changes will be minimal, whereas they will be large and of different sign at the edges (small and large mole fractions).
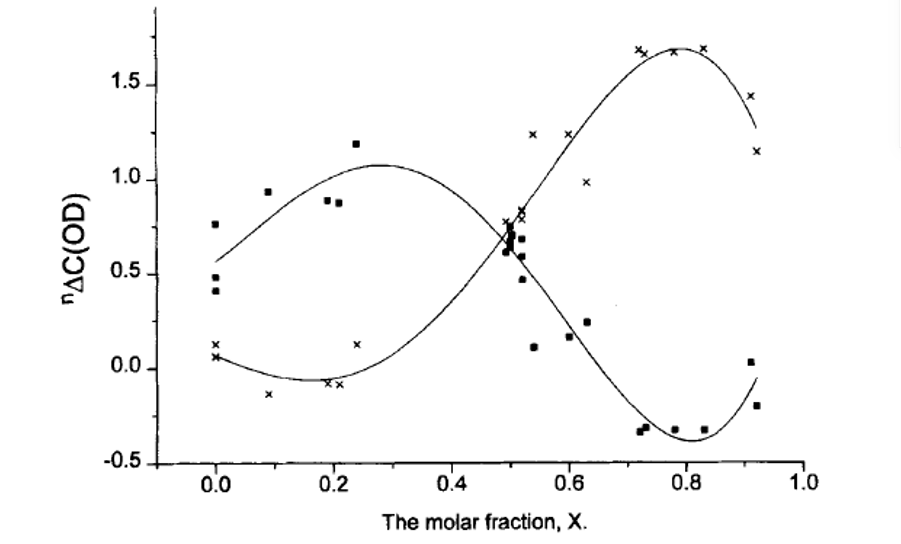
Figure 8. Plot of two-bond deuterium isotope effects of 13C chemical shifts vs. mole fraction. From Ref. 20 with Permission from Wiley-VCH.
Lifting degeneracy in symmetrical systems
13C NMR. Deuterium isotope effects on 13C chemical shifts can in a very elegant way be used to detect an equilibrium. The technique was originally invented in the study of carbonium ions. [21] A typical example of this technique is seen in Figure 9. The origin of the effect is the difference in CH stretching frequencies of the aldehyde, 2770 cm-1 and the enol type, 3020 cm-1.

Figure 9. Lifting of degeneracy by deuterium substitution. [22]
18O has been used as the isotope in order to distinguish between double well and symmetric hydrogen bond potentials in compounds having strong intramolecular hydrogen bonds between carboxylic acid and carboxylate groups. Figure 10. [23]
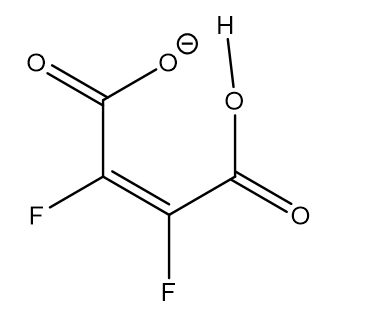
Figure 10. 18O labelled maleic anion.
What is the characteristics of equilibrium isotope effects? They can be measured at nuclei far from the site of isotope substitution and can be very large. In case of thioacetylacetone (Figure 4) deuteriation at the OH;SH position leads to 13C isotope effects at the “CS” carbon as large as -10.5 ppm depending on temperature. [2] Furthermore, as seen from Eq. 1 they will in those cases that they are larger than the intrinsic ones be proportional to the chemical shift difference between the nuclei in question (the chemical sift differences can be estimated from DFT calculations of NMR nuclear shieldings). An example of the latter is shown in Table 2.
Table 2. Calculated 13C nuclear shieldings for the Schiff base 2-(N-methyl-α-iminoethyl)-3,5-dibromophenol (Figure 6 with R= CH3 and R´= 3,5-dichloro)
|
|
C-a |
C-1 |
C-2 |
C-3 |
C-4 |
C-5 |
C-6 |
C-1´ |
Cst |
|
OH-form |
-11.0 |
13.9 |
-29.1 |
-9.0 |
-5.7 |
11.3 |
0.3 |
9.7 |
0.3 |
|
NH-form |
-28.9 |
5.5 |
9.3 |
13.5 |
0.3 |
1.0 |
0.8 |
-10.9 |
-1.9 |
From the above table we can predict that all carbons except C-4, C-6 and Cst will show large equilibrium isotope effects.
In addition, the ratio between isotope effects can be expressed in those cases in which the equilibrium isotope effects dominate:
ΔC-1/ ΔC-2 = (δC-1M- δC-1PT)/ (δC-2M- δC-2PT)
This shows the ratio between isotope effects for C1 and C2 of the two tautomers called M and PT.
An illustration is given in Figure 11 for a series of o-hydroxy Schiff bases (Figure 5). [7]
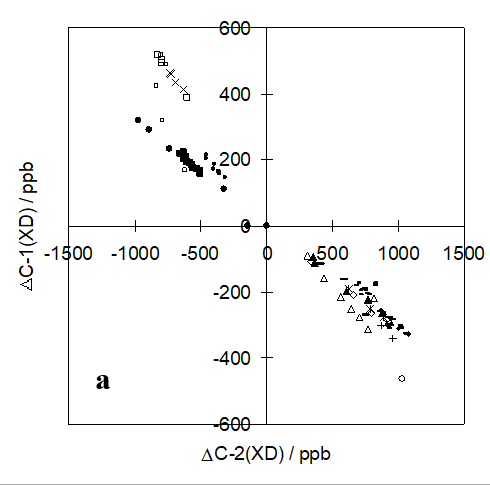
Figure 11. Ratios between deuterium isotope effects on 13C chemical shifts for C-1 and C-2 for a series of o-hydroxy Schiff bases. Taken from Ref. [7] with permission from the American Chemical Society.
Deuterium isotope effects on 19F chemical shifts
These have been demonstrated in o-hydroxyazo compounds (see Fig. 6). However, in the benzene derivative case no tautomerism is taking pIace ,the isotope effect is only 0.011 ppm, whereas in similar naphthalene derivatives the effects in the equilibrium case is 0.077 ppm or 0.123 ppm For the azo group in 1- respective 2-position. [24]
Deuterium isotope effects on 17O chemical shifts
These isotope effects are large at both oxygens involved (Figure 12). [9]
Figure 12. Deuterium isotope effects on 17O chemical shifts. Only one of the tautomers is shown.
Determination of equilibrium constants
As NMR parameters, chemical shifts and coupling constants, are average ones, the equilibrium constants can be determined, if we have determined the corresponding parameters for the two tautomers. Furthermore, as described in Figure 8 the mole fractions can be determined from deuterium isotope effects on 13C chemical shifts.
References
- Olivieri, A.C. Wilson, R.B. Paul, I.C. Curtin, D.Y; 13C NMR and X-ray Structure Determination of 1-(Arylazo)-2-naphthols. Intramolecular Proton Transfer between Nitrogen and Oxygen Atoms in the Solid State. J.Am.Chem.Soc. 1989, 111, 5525-5532.
- B. Andresen; F. Duus; S. Bolvig; P.E. Hansen; Variable temperature 1H and 13C NMR spectroscopic investigation of the enol–enethiol tautomerism of β-thioxoketones. Isotope effects due to deuteron chelation. Journal of Molecular Structure 2000, 552, 45-62, 10.1016/s0022-2860(00)00458-0.
- J.M Fernández-G; Federico Del Rio Portilla; B Quiroz-Garcı́a; Ruben Alfredo Toscano; R Salcedo; The structures of some ortho-hydroxy Schiff base ligands. Journal of Molecular Structure 2001, 561, 197-207, 10.1016/s0022-2860(00)00915-7.
- Antonin Lyčka; 19F-NMR study of azo-hydrazone tautomerism of some fluorine-containing azo dyes. Dyes and Pigments 1990, 12, 179-185, 10.1016/0143-7208(90)85010-l.
- Poul Erik Hansen; Alexsander S. Peregudov; Dimitrii N. Kravtsov; Antonina I. Krylova; Galina M. Babakhina; Ludmila S. Golovchenko; Valentina M. Pachevskaya; Prototropic processes in benzaurins. 19F and 1H NMR spectra of fluoro- and methylsubstituted 4-hydroxyphenyl-diphenylcarbinols, related fuchsones and benzaurins. Central European Journal of Chemistry 2011, 9, 253-264, 10.2478/s11532-011-0003-0.
- Kotovskaya, S.K. Romanova, S.A. Charushin, V.N.and Kodess, M.I; Fluorine-containing Heterocycles: XI. 5(6)-Fluoro-6(5)-X-benzofurans: synthesis, Tautomerism and Transformations. Russ.J.Org.Chem. 2004, 40, 1167-1174.
- A. Filarowski; A. Koll; M. Rospenk; I. Król-Starzomska; Poul Erik Hansen; Tautomerism of Sterically Hindered Schiff Bases. Deuterium Isotope Effects on13C Chemical Shifts. The Journal of Physical Chemistry A 2005, 109, 4464-4473, 10.1021/jp0445977.
- Boykin, D.W. 17O NMR Spectroscopy in Organic Chemistry. CRC Press, Boca Raton, Fl. 1991.
- S. Bolvig; Poul Erik Hansen; D. Wemmer; P. Williams; Deuterium isotope effects on 17 O chemical shifts of intramolecularly hydrogen bonded systems. Journal of Molecular Structure 1999, 509, 171-181, 10.1016/s0022-2860(99)00219-7.
- Hansen, P. E. Sitkowski J, Stefaniak, L. Rozwadowski, Z. Dziembowska, T; One Bond Deuterium Isotope Effects on 15N Chemical Shifts, 1DN(D), in Schiff Bases. Berichte Bunsengesel Chem.Phys. 1998, 102, 410-413.
- Hansen, P.E. Methods to distinguish tautomeric cases from static ones. In Tautomerism: Ideas, Compounds, Applications. Ed. L. Antonov. Wiley-VCH, 2016.
- Bordner, J. Hammen, P.D. and Whipple, E.B; Deuterium-Isotope Effects on C-13 NMR shifts and the Tautomeric Equilibrium in N-substituted Pyridyl Derivatives of Piroxicam. J.Am.Chem.Soc. 1989, 11, 6572-6578.
- Shasad Sharif; Gleb Denisov; Michael D. Toney; Hans-Heinrich Limbach; NMR Studies of Solvent-Assisted Proton Transfer in a Biologically Relevant Schiff Base: Toward a Distinction of Geometric and Equilibrium H-Bond Isotope Effects. Journal of the American Chemical Society 2006, 128, 3375-3387, 10.1021/ja056251v.
- Hans-Ullrich Siehl; Isotope Effects on nmr Spectra of Equilibrating Systems. Advances in Physical Organic Chemistry 1987, 23, 63-163, 10.1016/s0065-3160(08)60203-8.
- Forsyth, D.A. Isotopes in Organic Chemistry; Buncel, E.; Lee, C. C., Eds.; Elsevier: Amsterdam, 1987; Vol. 6, Chapter 1.
- Hansen, P.E; Deuterium Isotope Effects on Nuclear Shielding. Prog.Nucl.Magn.Reson. Spectrosc. 1988, 20, 207-257.
- Hansen, P.E. Isotope effects on chemical shifts as a tool in the study of tautomeric equilibria. In: Equilibria. Methods and theories. L.Antonov ed. Wiley-VCH. 2014.
- Dziembowska, T.; Rozwadowski, Z; Application of the deuterium isotope effect on NMR chemical shift to study proton transfer equilibria. Curr.Org. Chem. 2001, 5, 289-313.
- Guðmundur Gunnarsson; Håkan Wennerström; William Egan; Sture Forsén; Proton and deuterium NMR of hydrogen bonds: Relationship between isotope effects and the hydrogen bond potential. Chemical Physics Letters 1976, 38, 96-99, 10.1016/0009-2614(76)80264-3.
- Bolvig, S.; Hansen, P.E; Deuterium Isotope Effects on 13C Chemical Shifts as a Probe for Tautomerism in Enolic b-Diketones. Magn.Reson.Chem. 1996, 34, 467-478.
- Saunders, M. Telkowski, L. and Kates, M. R. J. Am. Chem. Soc. 1977, 99, 8070-8075.
- Charles L. Perrin; Yeong-Joon Kim; Symmetry of the Hydrogen Bond in Malonaldehyde Enol in Solution. Journal of the American Chemical Society 1998, 120, 12641-12645, 10.1021/ja9825579.
- Charles L. Perrin; Annadka Shrinidhi; Kathryn D. Burke; Isotopic-Perturbation NMR Study of Hydrogen-Bond Symmetry in Solution: Temperature Dependence and Comparison of OHO and ODO Hydrogen Bonds. Journal of the American Chemical Society 2019, 141, 17278-17286, 10.1021/jacs.9b08492.
- Poul Erik Hansen; Simon Bolvig; Ágnes Buvári-Barcza; Antonin Lyčka; José Maria Moreno; Antonio Romerosa; Ward T. Robinson; Björn O. Roos; Claire Vallance; Bryan R. Wood; et al. Long-Range Intrinsic and Equilibrium Deuterium Isotope Effects on 19F Chemical Shifts.. Acta Chemica Scandinavica 1997, 51, 881-888, 10.3891/acta.chem.scand.51-0881.