Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Dean Liu and Version 1 by Lidija Križančić Bombek.
The accumulation of oxidative damage to DNA and other biomolecules done by reactive oxygen species (ROS) plays an important role in the etiology of aging and age-related diseases such as type 2 diabetes mellitus (T2D), atherosclerosis, and neurodegenerative disorders. Mitochondrial DNA (mtDNA) is especially sensitive to oxidative stress. Mitochondrial dysfunction resulting from the accumulation of mtDNA damage impairs normal cellular function and leads to a bioenergetic crisis that accelerates aging and associated diseases.
- uncoupling proteins
- reactive oxygen species
- aging
- type 2 diabetes
1. Introduction
Mitochondria are the organelles of the cell that are responsible for energy production. Mitochondria are essential for aerobic ATP synthesis by oxidative phosphorylation and for the synthesis of heme, cholesterol, and phospholipids, as well as for apoptosis and cell signaling [1]. They are unique cell organelles because they have their own genome. Mitochondrial DNA (mtDNA) can self-replicate and transcribe. Because mtDNA is small and circular, it only encodes proteins essential for normal oxidative phosphorylation, namely, some subunits of the mitochondrial respiratory chain and some tRNA and rRNAs for the assembly of the mitochondrial translational machinery. The nuclear genome encodes all other proteins necessary for proper mitochondrial function, which are then imported into the mitochondria [2].
Mitochondria are also the largest source of reactive oxygen species (ROS) in the cell, which are generated when electrons leak during respiration [3]. At levels that are non-damaging, ROS are involved in important signal transduction pathways related to cell growth, apoptosis, kinase activation, immune responses, gene expression regulation, and calcium signaling [4,5,6,7,8,9,10][4][5][6][7][8][9][10]. However, excessive amounts of ROS not only directly damage lipids, proteins, and DNA, resulting in mtDNA mutations [11] but also affect a variety of stress-sensitive intracellular signaling pathways, such as the mitogen-activated protein kinase (MAPK) pathway, Jun amino-terminal kinase/stress-activated protein kinase (JNK/SAPK) pathway, and the nuclear factor kappa B (NF-kB) pathway [12,13,14,15,16][12][13][14][15][16]. Increased expression of the gene products of these pathways causes additional cellular damage [17,18][17][18]. mtDNA damage can impair viability and various cellular functions, and maintaining its integrity with age is crucial for survival [19]. Accordingly, mitochondrial dysfunction has been associated with various age-related diseases, such as type 2 diabetes (T2D), neurodegenerative diseases, cancer, and cardiovascular diseases [20,21,22,23,24][20][21][22][23][24].
T2D is a disease characterized by insufficient production of insulin, excessive secretion of glucagon by pancreatic beta cells, and insulin resistance, resulting in impaired energy metabolism in the pancreas, liver, skeletal muscle, and other organs [25]. Data for 2021 show that the global prevalence of T2D in 20- to 79-year-olds is 10.5%. The prevalence is lowest in young adults aged 20–24 years (2.2%) and steadily increases to 24% in elderly individuals aged 75–79 years. Projections for 2045 are similar, except that the percentages will be slightly higher in each age group. Most importantly, the aging of the world population will result in a higher proportion of people with T2D over the age of 60 [26[26][27],27], along with a higher incidence of cardiovascular complications and metabolic syndrome. The increased incidence of various comorbidities and the simultaneous use of different medications, which may lead to drug interactions in older diabetic patients, make the management of T2D particularly complex and challenging. Therefore, new approaches for controlling T2D are needed, including individualized treatment strategies [28].
Although the primary cause of T2D has not yet been determined, mitochondrial dysfunction in the organs responsible for insulin secretion (pancreatic beta cells), in the target organs of insulin action (skeletal and cardiac muscle cells and liver cells), and in the target organs associated with the major complications of T2D (kidneys, retina, nerves, and vascular cells) may play an important role in the pathophysiology of the disease [29]. Since ATP is critical for the production and release of insulin, altered mitochondrial bioenergetics associated with impaired glucose and fatty acid metabolism have been linked to defects in insulin and glucagon secretion in T2D [30].
UCPs are a group of five homologous proteins located in the inner mitochondrial membrane of various tissues. They are involved in several tasks and cellular functions, from thermoregulation to modulation of insulin secretion and neuroprotection [31,32,33,34][31][32][33][34]. The most diverse spectrum of UCPs is found in the mitochondria of skeletal muscle, which express all five UCPs. For this reason, skeletal muscle is one of the best-studied tissues in regard to advancing ourthe knowledge of UCP function and associated pathologies [35,36][35][36]. UCPs have been intensively studied in the last three decades because of their involvement in glucose and lipid metabolism [37,38,39,40,41,42,43][37][38][39][40][41][42][43]. In addition, many studies in mice, rats, and humans have shown that mitochondrial uncoupling proteins (UCPs) have important protective effects against oxidative stress and mitochondrial dysfunction [44,45,46][44][45][46]. However, their exact role has not been fully elucidated.
2. Mitochondria, ROS, and Oxidative Stress
The main source of ROS in the cell is the mitochondrial respiratory chain, which consists of four protein complexes responsible for generating the proton motive force across the inner mitochondrial membrane (Figure 1). Complex I (NADH-ubiquinone oxidoreductase) accepts electrons from NADH and passes them to complex II (succinate dehydrogenase), which oxidizes succinate to fumarate. As an enzyme of the Krebs cycle, complex II provides a direct link between the Krebs cycle and the respiratory chain [47]. Electrons from complexes I and II are transferred to ubiquinone (Q), which is then oxidized by complex III (ubiquinol cytochrome C oxidoreductase). Finally, electrons are passed to complex IV (cytochrome C oxidase) and used to reduce molecular O2 as the final electron acceptor, producing water. As electrons are transferred through the respiratory chain to complexes I, III, and IV, protons from NADH and FADH2 are translocated from the mitochondrial matrix into the intermembrane space, generating a strong proton motive force that subsequently drives the mitochondrial ATPase to produce ATP [3,47][3][47].
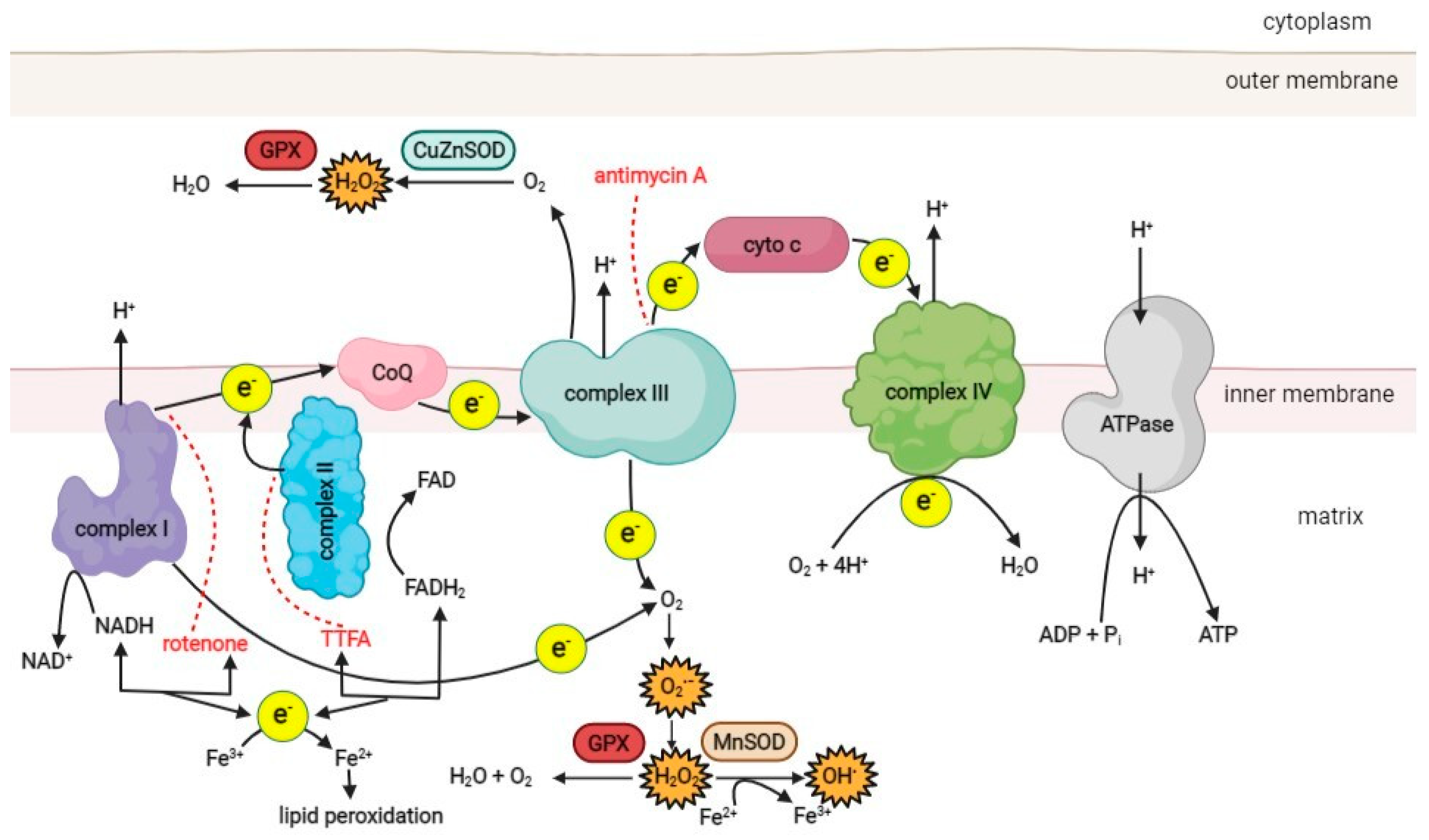
Figure 1. A schematic overview of the ROS production pathways in the respiratory chain of the mitochondrion. (CoQ–coenzyme ubiquinol, cyto c–cytochrome c, MnSOD–manganese superoxide dismutase, CuZnSOD–copper-zinc superoxide dismutase, GPX–glutathione peroxidase, TTFA–thenoyltrifluoroacetone). Red dotted lines represent the inhibition effect of specific compounds on the respiratory chain complexes. Created with BioRender.com.
Under physiological conditions, 1–5% of the oxygen consumed by the mitochondria is incompletely reduced to superoxide (O2•−), the primary ROS species formed in mitochondria, mainly in complexes I and III [48,49][48][49]. Other sources of ROS in the cell include NAD(P)H oxidase, various isoforms of nitric oxide synthase (NOS), xanthine oxidase, and lipoxygenases [6,50][6][50].
Superoxide is a charged molecule and, as such, does not readily diffuse across membranes. However, mitochondrial ROS can enter the cytosol after conversion to hydrogen peroxide (H2O2) by superoxide dismutase (SOD) [19]. There are three known SOD isoforms. SOD 1 (copper-zinc SOD; CuZn-SOD) is located in the mitochondrial intermembrane space, cytosol, and nucleus. SOD 2 (manganese SOD; Mn-SOD) is found only in the mitochondrial matrix, while SOD 3 (extracellular CuZn-SOD; EC-SOD) is present in the extracellular space [51]. In the mitochondrial matrix, H2O2 is reduced to water by catalase and glutathione peroxidase [52,53][52][53]. However, in the presence of transition metals such as copper or iron, H2O2 can be converted to reactive and damaging hydroxyl radicals (•OH) via the Fenton reaction or the Haber–Weiss reaction [6]. The resulting ROS can damage the proteins, lipids, and DNA of the cell. ROS generation, oxidative damage, and antioxidant defense mechanisms of the cell have been discussed in detail elsewhere [11,14,54][11][14][54].
Mitochondrial dysfunction, such as that associated with electron transport blockade, causes the respiratory chain to enter a highly reduced state. This triggers increased electron leakage and the production of superoxide anions and other ROS that further damage the cell’s biomolecules in a destructive cycle that can lead to progressive cell function degeneration and, eventually, cell death.
Mitochondria are not only the main producers of ROS but also their main target. In differentiated, nondividing cells, mtDNA is constantly replicating as intracellular ROS generation progresses. Oxidative stress in the form of various oxygen radicals modifies DNA. The damage leads to single- and double-strand breaks and base changes, resulting in cellular dysfunction, mutagenesis, and even carcinogenesis [19]. In particular, hydroxyl radicals are known to attack guanine bases [55]. One of the most common DNA lesions caused by ROS-induced mutagenesis is the modified guanine base 8-oxoguanine, which pairs equally efficiently with adenine and cytosine [19] and causes transversion mutations.
Since mitochondrial ROS production is much higher than that in the cytoplasm, ROS-induced damage to mtDNA is much more significant than damage to nuclear DNA. The mutation rate of mtDNA is up to twenty times higher than that of nuclear DNA, and its point mutation rate is more than two orders of magnitude higher than that of nuclear genes [56,57][56][57]. In addition, mitochondria tend to accumulate toxic xenobiotics. The matrix side of the mitochondrial membrane has a negative potential. It attracts lipophilic cations, including drugs and biotoxic chemicals, and causes their massive concentration, leading to exogenously induced mitochondrial damage [58,59,60][58][59][60]. Mutations in mtDNA accumulate with age and can lead to cellular dysfunction [19,61,62][19][61][62]. Large mtDNA deletions have been detected in healthy elderly humans and other species, such as Caenorhabditis elegans, mice, rats, and monkeys [63,64,65,66,67][63][64][65][66][67]. Moreover, an increased frequency of mitochondrial genomic deletions in brain samples has been associated with Huntington’s disease and Alzheimer’s disease [68,69][68][69].
Cells use various antioxidant systems to degrade ROS. One of the most important antioxidant enzymes in mitochondria is glutathione peroxidase [6,70][6][70]. Its function is to remove hydrogen peroxide, which is formed from superoxide anions (Figure 1). In addition, vitamin E, present in the inner mitochondrial membrane, acts as an antioxidant by accepting unpaired electrons and generating a stable product [71]. The oxidative damage repair system in mitochondria plays an important role in normal cellular function. It includes enzymes that repair oxidized mtDNA, eliminate mutant dNTPs, and degrade damaged mtDNA [72,73,74][72][73][74]. In humans, the MTH1 gene encodes 8-oxo-dGTPase, a human counterpart of the well-studied Escherichia coli protein MutT, which is essential for the removal of adenine paired with 8-oxoguanine in DNA [75]. Studies on the accuracy of mitochondrial DNA polymerase gamma in mtDNA replication and proofreading have shown that it is comparable to nuclear DNA polymerase [76], suggesting that higher mtDNA mutation rates result from more severe damage or/and weaker post-replication repair activities. Since dNTPs for mtDNA synthesis are synthesized inside mitochondria, all oxidized dNTPs must be removed in situ.
In addition to naturally occurring enzymatic and non-enzymatic antioxidants, mitochondria have endogenously regulated proteins called uncoupling proteins (UCPs) that can limit oxidative damage to cells.
3. Mitochondrial Dysfunction in T2D
Blood glucose levels must be adequately regulated to meet the energy needs of tissues while preventing excessive blood glucose levels from damaging blood vessel walls and nervous system cells. Blood glucose levels are controlled by two types of pancreatic islet cells: beta cells, which secrete insulin and amylin, and alpha cells, which secrete glucagon [77]. Insulin primarily causes cells to take up glucose from the blood and store it as glycogen or fat. Insulin also inhibits the mobilization of glucose from glycogen, protein, and fat stores [78]. Amylin released by beta cells inhibits alpha cells from producing glucagon [79]. Amylin and insulin are released by beta cells when blood glucose levels are high and inhibit the production of glucagon. Conversely, a fall in blood glucose levels causes the production of insulin and amylin by beta cells to be reduced, allowing alpha cells to produce glucagon unimpeded. The hormone glucagon increases blood glucose levels by causing the liver to break down glycogen stores and stimulating the formation of glucose from other small molecules through gluconeogenesis [79].
T2D is characterized by impaired pancreatic beta cell function and insulin resistance [80]. To maintain normal plasma glucose levels, the pancreas secretes more insulin in the early stages of the disease due to insulin insensitivity of peripheral tissues. As the disease progresses and pancreatic function deteriorates, insulin can no longer maintain glucose at a homeostatic level. As a result of the decreased responsiveness of the liver to insulin and abnormalities in the regulation of glucagon secretion, hepatic glucose production increases [81]. Along with decreased glycogen uptake and impaired insulin secretion, these events lead to hyperglycemia. As tissues become resistant to insulin, the pancreas compensates by producing more insulin, resulting in hyperinsulinemia. Another metabolic dysfunction that accompanies T2D is dyslipidemia, a condition characterized by abnormal lipid levels in the blood and a major risk factor for cardiovascular disease in T2D patients. Several processes are involved in T2D-associated dyslipidemia, including hyperglycemia, impaired lipid metabolism, and increased triglyceride synthesis as a result of insulin resistance [82]. Together, hyperglycemia, hyperinsulinemia, and dyslipidemia are important contributors to the increased oxidative stress associated with T2D and related pathologies [83].
Superoxide production rate depends on the concentration of potential electron donors and the local O2 concentration. In isolated mitochondria, significant O2•− production was observed under two conditions. First, when ATP production was low with consequently high proton motive force and a reduced coenzyme Q (CoQ) content; second, when the NADH/NAD+ ratio in the mitochondrial matrix was high [3]. The latter is particularly prominent in intense lipid or glucose metabolism, resulting in subsequent ROS generation and chronic diabetic complications [84].
Increased oxidative stress plays an important role in the onset and progression of T2D, as evidenced by increased levels of oxidative stress markers and reduced antioxidant levels in diabetic subjects [85]. Several mechanisms contribute to oxidative stress under diabetic conditions. These include disruption of the mitochondrial electron transport chain [86], increased activity of the polyol pathway [87], glucose autooxidation [88], and formation of advanced glycation end products (AGEs) [89]. Interestingly, in T2D patients and their first-degree relatives, serum levels of copper and iron, two potent prooxidant trace elements, have been found to be elevated and correlate with increased glycated hemoglobin levels [90]. Copper has the potential to increase the formation of ROS during the conversion of Cu(I) to Cu(II) [91]. In addition, under hyperglycemic conditions, iron and copper participate in glucose autooxidation that yields hydrogen peroxide, which undergoes further metal-catalyzed conversion to form the highly reactive hydroxyl radical [92]. Increased production of ROS and ROS-related cellular damage can also result from high-dose pharmaceutical iron supplementation, such as in anemic pregnant women, leading to gestational diabetes [93]. To combat excess ROS generation, cells produce antioxidants, as evidenced by their increased levels in blood and saliva samples from diabetic patients [94].
By promoting insulin resistance, impaired glucose tolerance, and mitochondrial dysfunction, oxidative stress further contributes to the progression of diabetes and associated pathologies.
References
- Pfanner, N.; Warscheid, B.; Wiedemann, N. Mitochondrial proteins: From biogenesis to functional networks. Nat. Rev. Mol. Cell. Biol. 2019, 20, 267–284.
- Bonawitz, N.D.; Clayton, D.A.; Shadel, G.S. Initiation and beyond: Multiple functions of the human mitochondrial transcription machinery. Mol. Cell 2006, 24, 813–825.
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13.
- Irani, K. Oxidant signaling in vascular cell growth, death, and survival: A review of the roles of reactive oxygen species in smooth muscle and endothelial cell mitogenic and apoptotic signaling. Circ. Res. 2000, 87, 179–183.
- Lo, Y.Y.; Wong, J.M.; Cruz, T.F. Reactive oxygen species mediate cytokine activation of c-Jun NH2-terminal kinases. J. Biol. Chem. 1996, 271, 15703–15707.
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84.
- Bedard, K.; Krause, K.-H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313.
- Schoonbroodt, S.; Piette, J. Oxidative stress interference with the nuclear factor-κB activation pathways. Biochem. Pharmacol. 2000, 60, 1075–1083.
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15.
- Görlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271.
- Juan, C.A.; de la Lastra, J.M.P.; Plou, F.J.; Pérez-Lebeña, E. The Chemistry of Reactive Oxygen Species (ROS) Revisited: Outlining Their Role in Biological Macromolecules (DNA, Lipids and Proteins) and Induced Pathologies. Int. J. Mol. Sci. 2021, 22, 4642.
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Mechanistic Insight into Oxidative Stress-Triggered Signaling Pathways and Type 2 Diabetes. Molecules 2022, 27, 950.
- Kamata, H.; Honda, S.; Maeda, S.; Chang, L.; Hirata, H.; Karin, M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 2005, 120, 649–661.
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462.
- Reynaert, N.L.; van der Vliet, A.; Guala, A.S.; McGovern, T.; Hristova, M.; Pantano, C.; Heintz, N.H.; Heim, J.; Ho, Y.S.; Matthews, D.E.; et al. Dynamic redox control of NF-kappaB through glutaredoxin-regulated S-glutathionylation of inhibitory kappaB kinase beta. Proc. Natl. Acad. Sci. USA 2006, 103, 13086–13091.
- Han, D.; Ybanez, M.D.; Ahmadi, S.; Yeh, K.; Kaplowitz, N. Redox regulation of tumor necrosis factor signaling. Antioxid. Redox Signal. 2009, 11, 2245–2263.
- Newsholme, P.; Gaudel, C.; Krause, M. Mitochondria and diabetes. An intriguing pathogenetic role. Adv. Mitochondrial Med. 2012, 942, 235–247.
- Evans, J.L.; Goldfine, I.D.; Maddux, B.A.; Grodsky, G.M. Are oxidative stress-activated signaling pathways mediators of insulin resistance and beta-cell dysfunction? Diabetes 2003, 52, 1–8.
- Kang, D.; Hamasaki, N. Alterations of mitochondrial DNA in common diseases and disease states: Aging, neurodegeneration, heart failure, diabetes and cancer. Curr. Med. Chem. 2005, 12, 429–441.
- Grubelnik, V.; Markovič, R.; Lipovšek, S.; Leitinger, G.; Gosak, M.; Dolenšek, J.; Valladolid-Acebes, I.; Berggren, P.-O.; Stožer, A.; Perc, M. Modelling of dysregulated glucagon secretion in type 2 diabetes by considering mitochondrial alterations in pancreatic α-cells. R. Soc. Open Sci. 2020, 7, 191171.
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070.
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795.
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159.
- Wallace, D.C. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: A dawn for evolutionary medicine. Annu. Rev. Genet. 2005, 39, 359–407.
- Zheng, Y.; Ley, S.H.; Hu, F.B. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat. Rev. Endocrinol. 2018, 14, 88–98.
- Sun, H.; Saeedi, P.; Karuranga, S.; Pinkepank, M.; Ogurtsova, K.; Duncan, B.B.; Stein, C.; Basit, A.; Chan, J.C.; Mbanya, J.C. IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res. Clin. Pract. 2022, 183, 109119.
- Atlas, D. International diabetes federation. In IDF Diabetes Atlas, 7th ed.; International Diabetes Federation: Brussels, Belgium, 2015.
- Longo, M.; Bellastella, G.; Maiorino, M.I.; Meier, J.J.; Esposito, K.; Giugliano, D. Diabetes and aging: From treatment goals to pharmacologic therapy. Front. Endocrinol. 2019, 10, 45.
- Patti, M.E.; Corvera, S. The role of mitochondria in the pathogenesis of type 2 diabetes. Endocr. Rev. 2010, 31, 364–395.
- Grubelnik, V.; Zmazek, J.; Markovič, R.; Gosak, M.; Marhl, M. Mitochondrial dysfunction in pancreatic alpha and beta cells associated with type 2 diabetes mellitus. Life 2020, 10, 348.
- Krauss, S.; Zhang, C.-Y.; Lowell, B.B. The mitochondrial uncoupling-protein homologues. Nat. Rev. Mol. Cell Biol. 2005, 6, 248–261.
- Rose, G.; Crocco, P.; De Rango, F.; Montesanto, A.; Passarino, G. Further support to the uncoupling-to-survive theory: The genetic variation of human UCP genes is associated with longevity. PLoS ONE 2011, 6, e29650.
- Ricquier, D.; Bouillaud, F. Mitochondrial uncoupling proteins: From mitochondria to the regulation of energy balance. J. Physiol. 2000, 529 Pt 1, 3–10.
- Rousset, S.; Alves-Guerra, M.C.; Mozo, J.; Miroux, B.; Cassard-Doulcier, A.M.; Bouillaud, F.; Ricquier, D. The biology of mitochondrial uncoupling proteins. Diabetes 2004, 53 (Suppl. S1), S130–S135.
- Križančić Bombek, L.; Čater, M. Skeletal Muscle Uncoupling Proteins in Mice Models of Obesity. Metabolites 2022, 12, 259.
- Van Den Berg, S.A.; Van Marken Lichtenbelt, W.; Willems Van Dijk, K.; Schrauwen, P. Skeletal muscle mitochondrial uncoupling, adaptive thermogenesis and energy expenditure. Curr. Opin. Clin. Nutr. Metab. Care 2011, 14, 243–249.
- Liu, J.; Li, J.; Li, W.-J.; Wang, C.-M. The role of uncoupling proteins in diabetes mellitus. J. Diabetes Res. 2013, 2013, 1–7.
- Kopecký, J.; Rossmeisl, M.; Flachs, P.; Bardová, K.; Brauner, P. Mitochondrial uncoupling and lipid metabolism in adipocytes. Biochem. Soc. Trans. 2001, 29, 791–797.
- Busiello, R.A.; Savarese, S.; Lombardi, A. Mitochondrial uncoupling proteins and energy metabolism. Front. Physiol. 2015, 6, 36.
- Schrauwen, P.; Hesselink, M.K. The role of uncoupling protein 3 in fatty acid metabolism: Protection against lipotoxicity? Proc. Nutr. Soc. 2004, 63, 287–292.
- Erlanson-Albertsson, C. The role of uncoupling proteins in the regulation of metabolism. Acta Physiol. Scand. 2003, 178, 405–412.
- Maurer, S.F.; Fromme, T.; Mocek, S.; Zimmermann, A.; Klingenspor, M. Uncoupling protein 1 and the capacity for nonshivering thermogenesis are components of the glucose homeostatic system. Am. J. Physiol. Endocrinol. Metab. 2020, 318, E198–E215.
- Saleh, M.C.; Wheeler, M.B.; Chan, C.B. Uncoupling protein-2: Evidence for its function as a metabolic regulator. Diabetologia 2002, 45, 174–187.
- Hirschenson, J.; Melgar-Bermudez, E.; Mailloux, R.J. The Uncoupling Proteins: A Systematic Review on the Mechanism Used in the Prevention of Oxidative Stress. Antioxidants 2022, 11, 322.
- Ježek, P.; Holendová, B.; Garlid, K.D.; Jabůrek, M. Mitochondrial uncoupling proteins: Subtle regulators of cellular redox signaling. Antioxid. Redox Signal. 2018, 29, 667–714.
- Mailloux, R.J.; Harper, M.-E. Uncoupling proteins and the control of mitochondrial reactive oxygen species production. Free Radic. Biol. Med. 2011, 51, 1106–1115.
- Mailloux, R.J. Teaching the fundamentals of electron transfer reactions in mitochondria and the production and detection of reactive oxygen species. Redox Biol. 2015, 4, 381–398.
- Brand, M.D. The sites and topology of mitochondrial superoxide production. Exp. Gerontol. 2010, 45, 466–472.
- Dröse, S.; Brandt, U. Molecular mechanisms of superoxide production by the mitochondrial respiratory chain. Adv. Exp. Med. Biol. 2012, 748, 145–169.
- Robertson, R.P.; Harmon, J.; Tran, P.O.; Tanaka, Y.; Takahashi, H. Glucose toxicity in β-cells: Type 2 diabetes, good radicals gone bad, and the glutathione connection. Diabetes 2003, 52, 581–587.
- Okado-Matsumoto, A.; Fridovich, I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu, Zn-SOD in mitochondria. J. Biol. Chem. 2001, 276, 38388–38393.
- Suttorp, N.; Toepfer, W.; Roka, L. Antioxidant defense mechanisms of endothelial cells: Glutathione redox cycle versus catalase. Am. J. Physiol.-Cell Physiol. 1986, 251, C671–C680.
- Salvi, M.; Battaglia, V.; Brunati, A.M.; La Rocca, N.; Tibaldi, E.; Pietrangeli, P.; Marcocci, L.; Mondovì, B.; Rossi, C.A.; Toninello, A. Catalase takes part in rat liver mitochondria oxidative stress defense. J. Biol. Chem. 2007, 282, 24407–24415.
- Cross, C.E.; Halliwell, B.; Borish, E.T.; Pryor, W.A.; Ames, B.N.; Saul, R.L.; McCord, J.M.; Harman, D. Oxygen radicals and human disease. Ann. Intern. Med. 1987, 107, 526–545.
- Cadet, J.; Douki, T.; Ravanat, J.L. Oxidatively generated damage to the guanine moiety of DNA: Mechanistic aspects and formation in cells. Acc. Chem. Res. 2008, 41, 1075–1083.
- Khrapko, K.; Coller, H.A.; André, P.C.; Li, X.-C.; Hanekamp, J.S.; Thilly, W.G. Mitochondrial mutational spectra in human cells and tissues. Proc. Natl. Acad. Sci. USA 1997, 94, 13798–13803.
- Taylor, R.W.; Barron, M.J.; Borthwick, G.M.; Gospel, A.; Chinnery, P.F.; Samuels, D.C.; Taylor, G.A.; Plusa, S.M.; Needham, S.J.; Greaves, L.C. Mitochondrial DNA mutations in human colonic crypt stem cells. J. Clin. Investig. 2003, 112, 1351–1360.
- Singer, T.P.; Ramsay, R.R. Mechanism of the neurotoxicity of MPTP: An update. FEBS Lett. 1990, 274, 1–8.
- Meyer, J.N.; Leung, M.C.; Rooney, J.P.; Sendoel, A.; Hengartner, M.O.; Kisby, G.E.; Bess, A.S. Mitochondria as a target of environmental toxicants. Toxicol. Sci. 2013, 134, 1–17.
- Ross, M.F.; Da Ros, T.; Blaikie, F.H.; Prime, T.A.; Porteous, C.M.; Severina, I.I.; Skulachev, V.P.; Kjaergaard, H.G.; Smith, R.A.; Murphy, M.P. Accumulation of lipophilic dications by mitochondria and cells. Biochem. J. 2006, 400, 199–208.
- Kajander, O.A.; Rovio, A.T.; Majamaa, K.; Poulton, J.; Spelbrink, J.N.; Holt, I.J.; Karhunen, P.J.; Jacobs, H.T. Human mtDNA sublimons resemble rearranged mitochondrial genomes found in pathological states. Hum. Mol. Genet. 2000, 9, 2821–2835.
- Bandy, B.; Davison, A.J. Mitochondrial mutations may increase oxidative stress: Implications for carcinogenesis and aging? Free Radic. Biol. Med. 1990, 8, 523–539.
- Cortopassi, G.A.; Arnheim, N. Detection of a specific mitochondrial DNA deletion in tissues of older humans. Nucleic Acids Res. 1990, 18, 6927–6933.
- Melov, S.; Lithgow, G.; Fischer, D.; Tedesco, P.; Johnson, T. Increased frequency of deletions in the mitochondrial genome with age of Caenorhabditis elegans. Nucleic Acids Res. 1995, 23, 1419–1425.
- Tanhauser, S.M.; Laipis, P.J. Multiple Deletions Are Detectable in Mitochondrial DNA of Aging Mice (∗). J. Biol. Chem. 1995, 270, 24769–24775.
- Edris, W.; Burgett, B.; Stine, O.C.; Filburn, C.R. Detection and quantitation by competitive PCR of an age-associated increase in a 4.8-kb deletion in rat mitochondrial DNA. Mutat. Res./DNAging 1994, 316, 69–78.
- Lee, C.M.; Chung, S.S.; Kaczkowski, J.M.; Weindruch, R.; Aiken, J.M. Multiple mitochondrial DNA deletions associated with age in skeletal muscle of rhesus monkeys. J. Gerontol. 1993, 48, B201–B205.
- Horton, T.; Graham, B.; Corral-Debrinski, M.; Shoffner, J.; Kaufman, A.; Beal, M.; Wallace, D. Marked increase in mitochondrial DNA deletion levels in the cerebral cortex of Huntington’s disease patients. Neurology 1995, 45, 1879–1883.
- Corral-Debrinski, M.; Horton, T.; Lott, M.T.; Shoffner, J.M.; McKee, A.C.; Beal, M.F.; Graham, B.H.; Wallace, D.C. Marked changes in mitochondrial DNA deletion levels in Alzheimer brains. Genomics 1994, 23, 471–476.
- Murphy, M.P. Mitochondrial thiols in antioxidant protection and redox signaling: Distinct roles for glutathionylation and other thiol modifications. Antioxid. Redox Signal. 2012, 16, 476–495.
- Thomas, S.M.; Gebicki, J.M.; Dean, R.T. Radical initiated alpha-tocopherol depletion and lipid peroxidation in mitochondrial membranes. Biochim. Biophys. Acta 1989, 1002, 189–197.
- Kang, D.; Hamasaki, N. Maintenance of mitochondrial DNA integrity: Repair and degradation. Curr. Genet. 2002, 41, 311–322.
- Kazak, L.; Reyes, A.; Holt, I.J. Minimizing the damage: Repair pathways keep mitochondrial DNA intact. Nat. Rev. Mol. Cell Biol. 2012, 13, 659–671.
- Liu, P.; Demple, B. DNA repair in mammalian mitochondria: Much more than we thought? Environ. Mol. Mutagen. 2010, 51, 417–426.
- Sakumi, K.; Furuichi, M.; Tsuzuki, T.; Kakuma, T.; Kawabata, S.-I.; Maki, H.; Sekiguchi, M. Cloning and expression of cDNA for a human enzyme that hydrolyzes 8-oxo-dGTP, a mutagenic substrate for DNA synthesis. J. Biol. Chem. 1993, 268, 23524–23530.
- Longley, M.J.; Ropp, P.A.; Lim, S.E.; Copeland, W.C. Characterization of the native and recombinant catalytic subunit of human DNA polymerase γ: Identification of residues critical for exonuclease activity and dideoxynucleotide sensitivity. Biochemistry 1998, 37, 10529–10539.
- Herman, M.A.; Kahn, B.B. Glucose transport and sensing in the maintenance of glucose homeostasis and metabolic harmony. J. Clin. Invest. 2006, 116, 1767–1775.
- Plum, L.; Belgardt, B.F.; Brüning, J.C. Central insulin action in energy and glucose homeostasis. J. Clin. Invest. 2006, 116, 1761–1766.
- Woods, S.C.; Lutz, T.A.; Geary, N.; Langhans, W. Pancreatic signals controlling food intake; insulin, glucagon and amylin. Philos. Trans. R Soc. Lond. B Biol. Sci. 2006, 361, 1219–1235.
- Kahn, S.E. The relative contributions of insulin resistance and beta-cell dysfunction to the pathophysiology of Type 2 diabetes. Diabetologia 2003, 46, 3–19.
- Ramnanan, C.J.; Edgerton, D.S.; Kraft, G.; Cherrington, A.D. Physiologic action of glucagon on liver glucose metabolism. Diabetes Obes. Metab. 2011, 13 (Suppl. S1), 118–125.
- Goldberg, I.J. Clinical review 124: Diabetic dyslipidemia: Causes and consequences. J. Clin. Endocrinol. Metab. 2001, 86, 965–971.
- Baynes, J.W.; Thorpe, S.R. Role of oxidative stress in diabetic complications: A new perspective on an old paradigm. Diabetes 1999, 48, 1–9.
- Berthiaume, J.M.; Kurdys, J.G.; Muntean, D.M.; Rosca, M.G. Mitochondrial NAD+/NADH redox state and diabetic cardiomyopathy. Antioxid. Redox Signal. 2019, 30, 375–398.
- Tiwari, B.K.; Pandey, K.B.; Abidi, A.B.; Rizvi, S.I. Markers of Oxidative Stress during Diabetes Mellitus. J. Biomark. 2013, 2013, 378790.
- Nishikawa, T.; Edelstein, D.; Du, X.L.; Yamagishi, S.-i.; Matsumura, T.; Kaneda, Y.; Yorek, M.A.; Beebe, D.; Oates, P.J.; Hammes, H.-P. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000, 404, 787–790.
- Chung, S.S.; Ho, E.C.; Lam, K.S.; Chung, S.K. Contribution of polyol pathway to diabetes-induced oxidative stress. J. Am. Soc. Nephrol. 2003, 14, S233–S236.
- Wolff, S.P.; Dean, R.T. Glucose autoxidation and protein modification. The potential role of ‘autoxidative glycosylation’ in diabetes. Biochem. J. 1987, 245, 243–250.
- Singh, V.P.; Bali, A.; Singh, N.; Jaggi, A.S. Advanced glycation end products and diabetic complications. Korean J. Physiol. Pharmacol. 2014, 18, 1–14.
- Atari-Hajipirloo, S.; Valizadeh, N.; Khadem-Ansari, M.H.; Rasmi, Y.; Kherad, A.F. Altered concentrations of copper, zinc, and iron are associated with increased levels of glycated hemoglobin in patients with type 2 diabetes mellitus and their first-degree relatives. Int. J. Endocrinol. Metab. 2016, 14, e33273.
- Lowe, J.; Taveira-da-Silva, R.; Hilario-Souza, E. Dissecting copper homeostasis in diabetes mellitus. IUBMB Life 2017, 69, 255–262.
- Harding, J.J.; Beswick, H.T. The possible contribution of glucose autoxidation to protein modification of diabetes. Biochem. J. 1988, 249, 617–618.
- Zhuang, T.; Han, H.; Yang, Z. Iron, oxidative stress and gestational diabetes. Nutrients 2014, 6, 3968–3980.
- Astaneie, F.; Afshari, M.; Mojtahedi, A.; Mostafalou, S.; Zamani, M.J.; Larijani, B.; Abdollahi, M. Total antioxidant capacity and levels of epidermal growth factor and nitric oxide in blood and saliva of insulin-dependent diabetic patients. Arch. Med. Res. 2005, 36, 376–381.
More