Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 3 by Jessie Wu and Version 5 by Jessie Wu.
Nonalcoholic fatty liver disease (NAFLD) is one of the most common causes of liver diseases, and its prevalence continues to increase worldwide. NAFLD is a spectrum of liver diseases that occur in the absence of other known causes, such as excess alcohol use. Since NAFLD is a metabolic disease, it has been recently renamed metabolic-associated fatty liver disease (MAFLD).
- non-alcoholic fatty liver disease (NAFLD)
- non-alcoholic steatohepatitis (NASH)
- metabolic associated fatty liver disease (MAFLD
- lipotoxicity
- fatty liver
1. Current Interventions in the Management of Nonalcoholic Fatty Liver Disease
1.1. Lifestyle Interventions
Lifestyle interventions have positively impacted nonalcoholic fatty liver disease (NAFLD) even in the absence of weight loss [1][2][3]. It is now recognized that a 5% weight reduction is associated with reduced liver fat and improved liver injury while body weight loss of >7% body weight improved NASH based on histology [1][4][5][6][7]. Importantly, weight loss percentage correlated with improved NASH histologic parameters [5]. While inflammation is essential for disease progression, the strongest predictor of mortality in patients with NASH is hepatic fibrosis [8][9]. Among patients who lost ~10% body weight, 90% showed improved NASH, and about ~45% showed fibrosis regression [9][10]. Lifestyle interventions involving a combination of calorie restriction and exercise have a higher impact on reducing liver fat [10]. However, more than 50% of patients included in clinical trials could not achieve this weight loss level [11]. Therefore, even though lifestyle interventions positively impacted NAFLD, sustained lifestyle changes are difficult to achieve [12].
Dietary interventions improve NAFLD with or without physical activity; however, the composition of the diet and the dietary pattern is still a matter of debate [5][7][11][13]. The picture is a little clearer with exercise interventions, as there is agreement that, in most clinical and preclinical studies, all exercise modalities and intensities are beneficial in NAFLD. Exercise has been shown to decrease hepatic steatosis, liver enzymes, blood glucose, and insulin and improve lipid profile, with or without dietary interventions [2][5][10][14]. Even without weight loss, regular exercise reduced hepatic lipids [15][16]. Exercise modulates FA synthesis and oxidation, mitochondria bioenergetics, and structure in the liver, thus preventing liver damage [15][17][18][19]. Exercise reduces hepatic fat by 1) down-regulating FA synthesis and upregulating FA oxidation, 2) decreasing oxidative stress and increasing antioxidant enzymes, and 3) reducing inflammation [20][21]. However, further research is needed to understand the underpinning mechanisms involved in the role of mitochondria and the epigenetics/posttranslational related mechanisms involved in the liver adaptation to lifestyle interventions. The 2018 ASSLD practice guidance states that weight loss reduces hepatic steatosis, achieved with hypocaloric diet, increased physical activity, or both. A combination of hypocaloric diet and moderate-intensity exercise is likely to result in sustained weight loss over time. Body weight loss of 3–5% improves steatosis, 7–10% body weight loss is needed to improve and including fibrosis [22].
1.2. Bariatric Surgery
Bariatric surgery is now recommended as an effective approach to treating clinically severe obesity and its complications [23][24]. Based on liver biopsy, bariatric surgery in morbidly obese patients improves steatosis, NASH, and liver fibrosis in 30% of patients, [25]. However, despite the significant benefit of bariatric surgery for resolving NASH, the risk of these surgeries currently excludes their use as first-line therapy for NAFL and NASH patients.
1.3. Pharmaceutical Therapies
As of now, no pharmacological treatment is approved to treat NAFLD. Better understanding of the pathogenesis of NAFLD has led to the investigation of potential drug molecules in clinical trials, to determine their efficacy and safety in the resolution of steatohepatitis and fibrosis. These drug molecules target several aspects of metabolic disruption including lipotoxicity, oxidative stress, mitochondrial dysfunction, and fibrosis. The drugs currently used for the treatment of NAFLD include off label treatments such antioxidant (VitE) and antidiabetic drugs such as pioglitazone. The farnesoid X receptor agonist obeticholic acid (OCA), the thyroid hormone receptor THRβ agonist (Resmetirom), and Aramchol (bile acid and and FA conjugate, cholic acid–arachidic acid), are drugs currently in phase 3 randomized clinical trials (RCTs) for the treatment of noncirrhotic NASH [26]. Drugs in phase 3 and several drugs in phase II phase 2 RCTs are reviewed in this section.
Vitamin E
Vitamin E, the bioactive α-tocopherol, is a potent antioxidant that helps maintain intracellular redox status by counteracting the increase in oxidative stress. In the PIVENS, phase 3 RCT, non-diabetic participants with NASH, based on liver biopsy, were administered daily treatments of 30 mg Pioglitazone, 800 IU VitE, or placebo for 96 weeks. Vitamin E therapy, as compared with placebo improved NASH. Both VitE and Pioglitazone significantly lowered ALT and AST levels, reduced hepatic steatosis and lobular inflammation but without improvement in fibrosis scores. The side effects were weight gain with Pioglitazone (ClinicalTrials.gov number, NCT00063622) [27]. In the TONIC trial, VitE (800 IU/day) or metformin 500 mg twice-daily were tested against placebo in children with biopsy-proven NAFLD [28]. VitE improved NASH compared to placebo [28]. The 2018 AASLD updated practice guidance stated that VitE administered at a of 800 IU/day may be considered for this patient population. However, VitE is not recommended to treat NASH in diabetic patients, NAFLD without liver biopsy, NASH cirrhosis, or cryptogenic cirrhosis [22]. Long-term safety of high-dose vitamin E need further evaluation for efficacy and safety. Metformin is not recommended for treating NASH in adult patients [22].
Pioglitazone, PPAR Agonist
PPAR targeting drugs, including thiazolidinediones such as Pioglitazone, are clinically used to treat T2D. In the PIVENS trial, treatment of nondiabetic NASH subjects with Pioglitazone (30mg/day) reduced hepatic steatosis, lobular inflammation, hepatocellular ballooning, improved insulin resistance and liver-enzyme levels, and reduced liver injury [27]. However, Pioglitazone treatment was associated with increased body weight gain [27]. Pioglitazone is contraindicated in patients with established heart failure or with an increased risk of heart failure [29]. Low dose of Pioglitazone (15mg/day) is being evaluated in a phase II clinical trial (Clinicaltrials.gov, NCT04501406) to evaluate the effect Pioglitazone on liver histology in patients with NASH. Clinical trials evaluating other PPAR agonists are ongoing [30]. The 2018 AASLD practice guidance states that Pioglitazone may be used to treat patients with and without T2D, with biopsy-proven NASH. Further pioglitazone efficacy and safety evaluation is needed. The 2018 ASSLD practice guidance states that Pioglitazone should not be used to treat NAFLD without biopsy-proven NASH [22].
Obeticholic Acid, Farnesoid X Receptor
Farnesoid X receptor FXR is a ligand-activated nuclear receptor transcription factor abundantly expressed in the liver, intestine, and kidney [31]. FXR regulates several metabolic functions, including bile acid synthesis, glucose homeostasis, and lipid metabolism [32]. Activation of FXR by ligands induces a small heterodimer partner, which suppresses CYP7A1 gene expression. CYP7A1 is a rate-limiting enzyme that converts cholesterol to bile acids, inhibition of which results in the reduced rate of bile synthesis in the liver [31]. Bile acids are FXR natural ligands. Activation of FXR in both hepatocytes and enterocytes reduced bile acid synthesis and improved hepatic steatosis and inflammation [32]. In preclinical studies, FXR agonists have shown beneficial effect on NASH [32]. In the FLINT study [33], FXR ligand obeticholic acid (OCA) was evaluated for the treatment of NASH (clinicaltrail.gov, NCT01265498). OCA improved NASH, fibrosis, and markers of hepatic damage [33]. REGENERATE is a phase 3 global RCT to evaluate the impact of OCA on NASH with fibrosis (clinicaltrials.gov, NCT02548351). Histologic assessment of patients with NASH and F2-F3 fibrosis demonstrated significant improvement in fibrosis by one stage with no resolution of NASH. The most adverse effect of OCA treatment are high rates of pruritus [34].
Resmetirom, Thyroid Hormone Receptor THRβ Agonists
Thyroid hormones (THs) regulate many processes involved in hepatic TG and cholesterol metabolism. Triiodothyronine (T3) is the major active form of THs and exerts its action by binding to two TH receptor, THRα and THRβ, which act as ligand-inducible transcription factors. THs increases influx of FAs in the liver by upregulating the expression of CD36, FABP and lipogenic genes [35]. However, THs increases TAG hydrolysis by stimulating the transcription and activities of ATGL, therefore mediating the mobilization of free FAs from TG stores and their subsequent β-oxidation [35]. THs also downregulate stearoyl-CoA desaturase-1 (SCD1), a key enzyme involved in triglyceride biosynthesis and GPAT, consequently limiting the storage of LDs in the liver [35]. THRβ is expressed in the liver while THRα is expressed in the heart and bone. Resmetirom is a selective THRβ agonist that has been developed to specifically activate THRβ in the liver and eliminate the side effects associated with the activation of THs in other tissues. Resmetirom was evaluated in adults with NASH (clinicaltrials.gov, NCT02912260) [36]. Compared to patients treated with placebo, Resmetirom reduced liver fat on MRI-PDFF, and reduced NASH [36]. Resmetirom treatment appears to be safe, the adverse effects are mild diarrhea and nausea. Resmetirom is being evaluated in phase III MAESTRO-NASH trial to assess its efficacy and safety of in patients with NASH and fibrosis (clinical trial.gov, NCT03900429).
Cholic Acid-Arachidic Acid Conjugate
Aramchol is a synthetic conjugate of bile acid (cholic acid) and FA (arachidic acid) that inhibits SCD-1. Treatment of methionine and choline deficient (MCD) mouse, a mouse model of NASH, with Aramchol improved NASH and fibrosis [37]. In the phase II 2b RCT, Aramchol was evaluated for its efficacy and safety versus placebo, in patients with NASH who had overweight or obesity and had confirmed prediabetes or T2D (ARREST, clinical trial NCT02279524). Aramchol (daily doses of 400 mg or 600 mg) for 52 weeks decreased liver fat and improved liver enzymes with a trend toward higher NASH improvement with the 600 mg dose [38]. Aramchol is currently being investigated in the ARMOR phase III clinical trial (clinical trial.gov, NCT04104321) to evaluate its efficacy and safety in patients with NASH and F2-F3 fibrosis.
Glucagon-Like Peptide Receptor Agonist
Glucagon-like peptide (GLP-1) and glucose-dependent insulinotropic peptide (GIP) are the two primary incretin hormones secreted by the L-cells and K-cells of the small intestine, respectively [39]. GIP and GLP-1 bind to their specific G-protein coupled receptors to initiate downstream signaling events in pancreatic β cells to stimulate glucose-dependent insulin secretion [39]. In a phase II study in patients with NASH and fibrosis, the GLP-1 receptor Semaglutide, improved NASH without worsening of fibrosis [40]. In the LEAN clinical trial, the GLP-1 receptor agonist Liraglutide, showed efficacy in reducing liver fat content as well as liver enzymes in patients with NASH (clinical trial.gov, NCT01237119). Liraglutide was safe, well tolerated, and led to histological resolution of NASH [41]. Liraglutide was associated with greater weight loss but also gastrointestinal side effects [41]. The SYNERGY-NASH, a phase II study to assess Tirzepatide, a dual GIP/GLP-1 receptor agonist in participants with NASH (clinicaltrials.gov, NCT04166773) is underway (NCT04166773). Phase II clinical trials using other agonists, or a combination of agonists are currently ongoing. The 2018 ASSLD practice guidance states GLP-1 agonists are not currently considered to specifically treat liver disease in patients with NAFLD or NASH [22].
Sodium-Glucose Co-Transporter Type 2 Inhibitor
Sodium-glucose co-transporter type 2 inhibitors (SGLT2i) are used as antidiabetic drugs. SGLT2 is almost exclusively expressed in the kidney and reabsorbs >90% of the glucose filtered at the glomerulus [42]. Pharmacological inhibition of SGLT2 using Dapagliflozin improved liver steatosis and attenuated liver fibrosis only in patients with significant liver fibrosis [43]. In patients with T2D and NAFLD, Dapagliflozin improved liver function parameters and decreased serum level of hepatocytes-secreted soluble dipeptidyl peptidase-4 (DPP4) which is responsible for adipose tissue inflammation and insulin resistance [43]. A phase III multi-center RCT is ongoing to assess the safety and the efficacy of Dapagliflozin in improving NASH (clinicaltrial.gov, NCT03723252). Other SGLT2i are being evaluated in phase II clinical trials.
Fibroblast Growth Factors Activators
Fibroblast growth factors (FGFs) are a superfamily of metabolic hormones that regulate many aspects of the whole-body health. Circulating FGF21 is liver-derived [44], but it is also expressed in several other tissues, such as the pancreas, muscle, and adipose. FGF19 and its mouse ortholog FGF15 [44] are gut-produced hormones with the highest expression in the ileum [44]. Both FGF19 and FGF21 play an important role in the liver [44][45]. FGF19 and FGF21 signal through FGF receptors in the body. The activity of FGF19 and FG21 requires a transmembrane scaffold protein bKlotho (KLB) [44]. FGF21 analogs have demonstrated efficacy in animal models and humans with NASH, and several clinical trials with FGF21 analogs are currently underway. Two FGF21 molecule in RCTs are reviewed here, Pegbefermin and Efruxifermin.
Pegbelfermin, FGF21 Analog
Pegbelfermin, a PEGylated, recombinant human FGF21 analog, was evaluated for its efficacy and safety in a phase II RCT. Subcutaneous treatment with Pegbelfermin, in obese/overweight subjects with NASH, for 16 weeks reduced liver fat measured by MRI-PDFF, improved biomarkers of metabolic function (adiponectin and lipid concentrations), and biomarkers of fibrosis. Pegbelfermin is being evaluated for its efficacy and safety in a two phase 2b RCT in patients with NASH and stage 3 fibrosis FALCON 1 (clinicaltrials.gov, NCT03486899), or in cirrhosis, FALCON 2 (clinicaltrials.gov, NCT03486912).
Efruxifermin, Fc-FGF21 Fusion Protein
BALANCED, a Phase 2a RCT in patients with histologically confirmed NASH, evaluated the safety and efficacy of fruxifermin, a long-acting Fc-FGF21 fusion protein, in a 16-week study (clinicaltrials.gov NCT03976401) [46]. The treatment with Efruxifermin was safe, except for diarrhea and nausea in approximately 30% of participants. Efruxifermin improved NAFLD activity score (NAS) and fibrosis, reduced body weight and liver fat content, and improved circulating TG and increased HDL.
mRNA Encoding Human FGF21
The therapeutic levels of FGF21 were achieved following subcutaneous administration of mRNA encoding human FGF21 proteins. FGF21 mRNA was assessed following 2-weeks repeated subcutaneous injections in diet-induced obese mice, which resulted in marked decreases in body weight, plasma insulin levels, and hepatic steatosis [47]. Studies in both lean and diet induced obesity mice showed that mRNA encoding human proteins provided better therapeutic coverage than recombinant proteins, in vivo suggesting that FGF 21 mRNA therapy might have the potential to treat T2D and NASH.
Statins
T2D is associated with increased risk of both cardiovascular disease (CVD), NASH and liver fibrosis. Satins are known to lower CVD. The effect of statins on liver fibrosis and hepatic steatosis were assessed in adult patients with T2D, in a cross-sectional study using data from the 2017–2018 cycle of the National Health and Nutrition Examination Survey (NHANES). Statins use was associated with lower odds of advanced fibrosis [48]. The updated ASSLD practice guidance indicates that statins can be used in the treatment of dyslipidemia in patients with NAFLD and NASH. While statins may be used in patients with NASH cirrhosis, they should be avoided in patients with decompensated cirrhosis [22]. Studies evaluating the safety and efficacy of statins in NASH and fibrosis are limited. STAT-NASH, a phase 2 RCT, is underway to assess the treatment of NASH with statins (clinicaltrials.gov, NCT04679376).
2. Sirtuins as Targets for Nonalcoholic Fatty Liver Disease Treatment
SIRTs represent potential targets for the treatment of NAFLD due to their role in hepatic lipid and carbohydrate metabolism, insulin signaling, redox signaling, and inflammation [49][50][51][52][53][54][55]. SIRTs are a family of seven members (SIRT1–7) with different cellular localization and are implicated in multiple cellular processes. SIRT1 and SIRT3 are NAD+-dependent deacetylases regulated by cellular NAD+/NADH ratio. SIRT1 and SIRT3 are upregulated by fasting, calorie restriction, exercise, and polyphenols and downregulated by nutrient overload. This section focuses on the most studied SIRTs, SIRT1 and SIRT3 (Figure 1 and Figure 2).
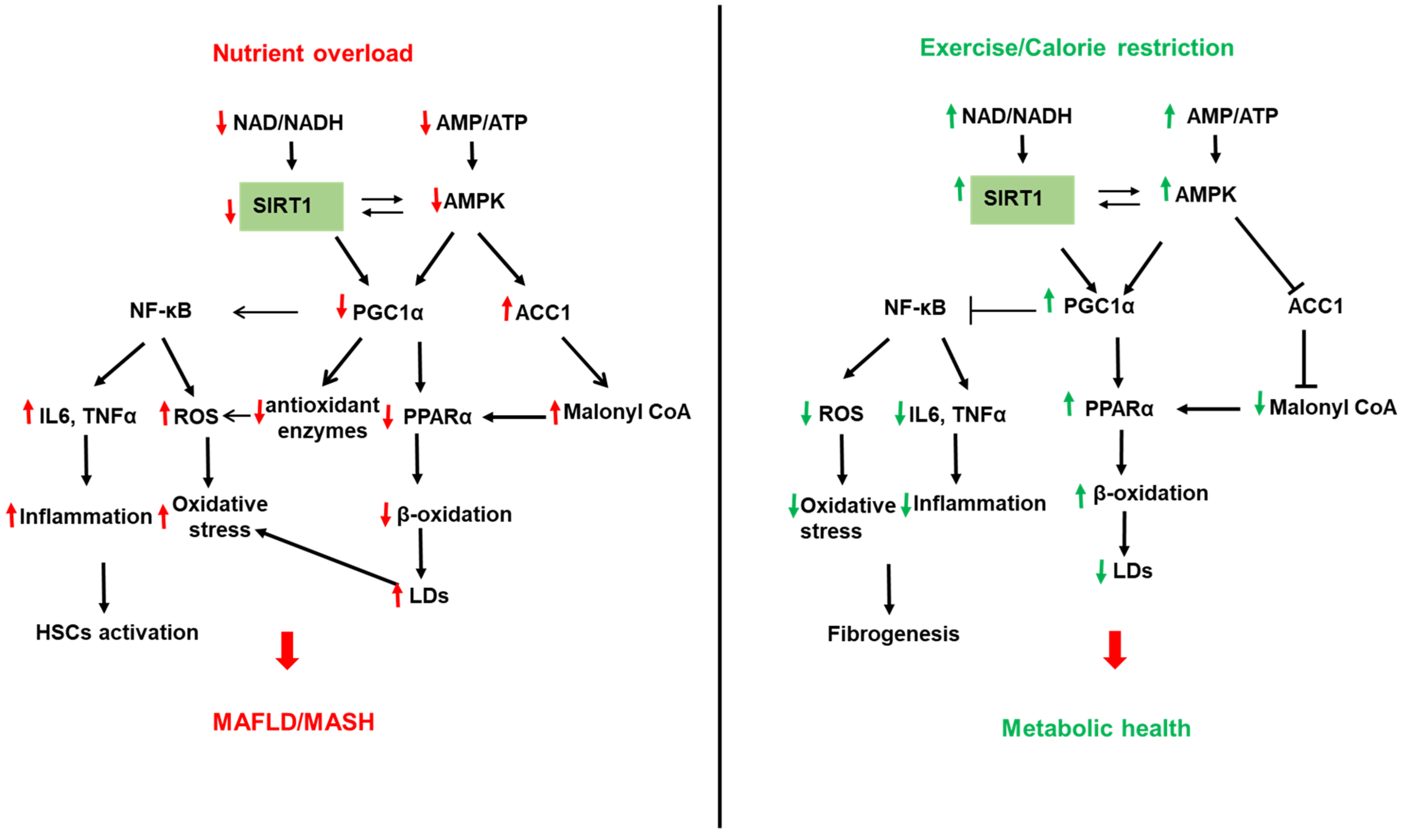
Figure 1. Role of sirtuin (SIRT) 1 in NAFLD. Left panel: Reduced SIRT1 with nutrient overload leads to the acetylation and inactivation of the peroxisome proliferator-activated receptor (PPAR) gamma-coactivator α (PGC1α). Reduced PGC1α activity downregulates β-oxidation by reducing PPARα expression and decreasing mitochondrial biogenesis, leading to increased lipid accumulation in LDs and hepatic steatosis. In addition, reduced SIRT1 activity decreases the expression of the antioxidant enzymes (SOD2 and catalase), leading to increased cellular ROS. On the other hand, reduced PGC1α activity increases the expression of the transcription factor NF-κB to increase inflammatory cytokines such as TNFα and IL6 and ROS generation. In addition, reduced AMP/ATP ratio with nutrient overload inactivates AMP-activated protein kinase (AMPK), leading to reduced NAD/ATP ratio and reduced SIRT1 and PGC1α activities. AMPK also directly regulates PGC1 via modulation of its phosphorylation. Reduced AMPK also reduces ACC1 phosphorylation and increases its activity leading to increased malonyl CoA and inhibition of β-oxidation. Right panel: Upregulation of SIRT1 by exercise, calorie restriction, and fasting upregulates SIRT1 leading to a healthier liver via opposite effects on B-oxidation, inflammation, and ROS generation than chronic nutrient overload.
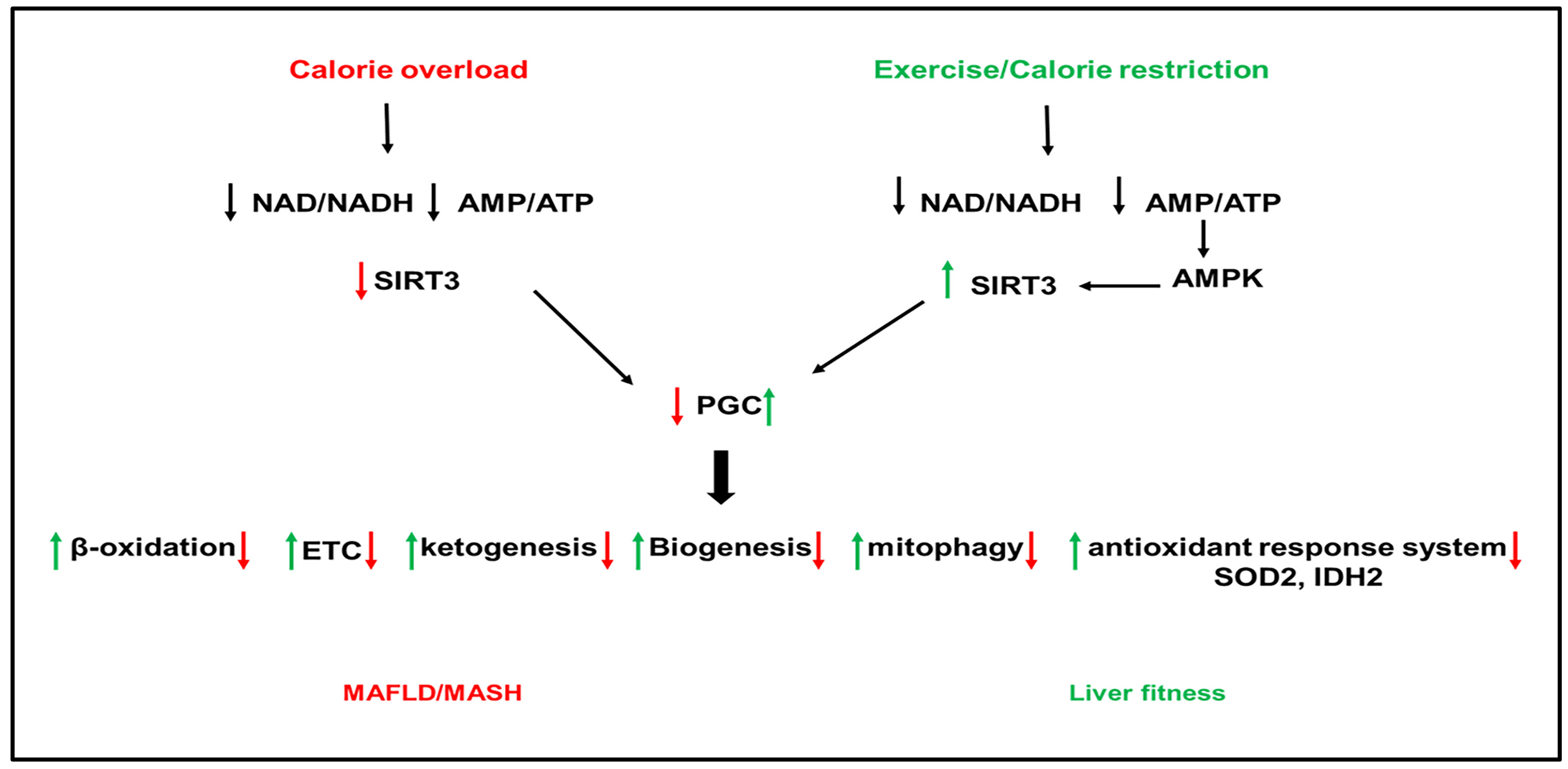
Figure 2. Role of sirtuins (SIRT) 3 in NAFLD. Calorie overload and liver lipotoxicity downregulate SIRT3 activity leading to the inactivation of PGC1α. The inactivation of PGC1α downregulates the downstream pathways, including β-oxidation, antioxidant enzymes (superoxide dismutase (SOD)2 and isocitrate dehydrogenase (IDAH)2, the ETC, ketogenesis, biogenesis, and mitophagy, leading to the development of hepatic steatosis, inflammation, and fibrogenesis. Activation of SIRT3 by exercise and calorie restriction activates PGC1α with opposite effects on PGC1 downstream pathways than nutrient overload and lipotoxicity, leading to metabolic health.
2.1. SIRT1 and NAFLD
SIRT1 is found in the nucleus and shuttles between the nucleus and cytoplasm under physiological and pathological conditions [55][56]. SIRT1 regulates, via deacetylation of transcription factors and proteins, multiple metabolic pathways in the liver, including FA synthesis and oxidation, oxidative phosphorylation, inflammation, mitochondrial biogenesis, and autophagy [51][55][57][58] (Figure 2). The involvement of SIRTs in NAFLD has been shown in both human and animal models of NAFLD. SIRT1 is downregulated in humans with NAFLD, and this was associated with increased expression of lipogenic proteins, such as SREBP1, ACC, and FAS [59]. Furthermore, the lack of SIRT1 catalytic activity promoted the release of free FAs from mesenteric adipose tissue and aggravated NAFLD [60]. SIRT1 levels were low in obese compared to lean patients and lower in obese patients with severe hepatic steatosis compared to obese patients with mild hepatic steatosis [61]. PPARγ coactivator-α (PGC1α) directly coactivates multiple transcription factors, including nuclear receptors such as PPARs, the thyroid hormone receptor, estrogen receptors, and estrogen-related receptors (ERRs). In addition, PGC1α coactivates transcription factors such as the family of forkhead O-box (FOXO) transcription factors [62]. PGC1α activity is regulated by expression levels and posttranslational modifications such as acetylation and phosphorylation. SIRT1 can directly deacetylate and activate PGC1α [63]. In addition, SIRT1 interacts with multiple transcriptional factors, resulting in enhanced β-oxidation and mitochondrial biogenesis [58]. PPARα is a transcription factor able to bind FAs and increase the expression of genes related to FA catabolism in the mitochondria. In fasting conditions, SIRT1 deacetylates PGC1α, which activates PPAR-α to promote FA oxidation and ATP production [64][65] (Figure 2). Interestingly, SIRT1 transgenic mice have similar phenotypes to mice on a calorie-restricted diet [66]. ATGL positively regulates SIRT1 deacetylase activity to promote PGC1α signaling. ATGL increases LDs lipolysis and the activity of the nuclear receptor PPARα to promote FA oxidation [64]. Liver-specific deletion of SIRT1 resulted in fatty liver, inflammation, and endoplasmic reticulum stress, due to impaired PPARα/PGC1α pathway [65][67]. SIRT1/PGC1α pathway mediates the beneficial effect of antioxidant treatment on mitochondrial function and oxidative stress in hepatocytes [68]. PGC1α increases the expression of ROS detoxifying enzymes such as SOD2, catalase, and antioxidant treatment improves oxidative stress caused by excess fructose via upregulation of SIRT-1 expression [56]. PGC1α, SIRT1, and AMPK represent an energy sensing network that controls metabolic homeostasis [69] (Figure 2). In addition to AMPK, cGMP, endothelial NO synthase, and exogenous NO are all upstream mediators of PGC1α and can increase mitochondrial biogenesis via PGC1α activation [70]. In high energy demands conditions, AMPK is activated by a high AMP/ATP ratio [71]. Once activated, AMPK turns on catabolic pathways to produce ATP while simultaneously turning off energy-consuming anabolic processes. To perform these actions, AMPK quickly regulates metabolic enzymes through direct phosphorylation, but additionally, AMPK can enhance SIRT1 activity by increasing cellular NAD+ levels, resulting in the deacetylation of PGC1α. Indeed, AMPK and SIRT1 share common target molecules, including PGC1α, PPARγ, and NF-κB [72][73][74]. Moreover, the treatment of mice fed HFD with resveratrol, a SIRT1 activator, activated the AMPKα-SIRT1 pathway, improved hepatic steatosis, and decreased inflammation [75]. The nicotinamide phosphoribosyltransferase (NAMPT) is a rate-limiting enzyme in NAD+ biosynthesis that regulates the activity of NAD+-dependent enzymes, such as SIRTs. SIRT1 mediates NAMPT’s effects on lipid metabolism and inflammation [76][77][78][79]. Inhibition of NAMPT aggravated the HFD-induced hepatic steatosis by suppressing the SIRT1-mediated signaling pathway [80]. NAD+ precursors improved hepatic mitochondrial function and decreased oxidative stress in Pre-clinical NAFLD models [75]. NAD+ repletion, using NAD+ precursors such as nicotinamide riboside and nicotinamide mononucleotide, reduced the activation of HSCs and prevented fibrosis and NASH progression. However, initial clinical trials have only shown modest effects when NAD+ precursors in obesity [78].
2.2. SIRT3 and Nonalcoholic AFatty Liver LDisease
SIRT3 is a mitochondrial NAD+-dependent deacetylase that regulates the activity of proteins involved in cellular metabolism [50][81]. SIRT3 gene expresses three isoforms, the two long isoforms of murine SIRT3 proteins (M1 and M2) are in the mitochondria. In contrast, the short form of SIRT3 protein (M3) lacks an N-terminal mitochondrial targeting signal in the cytosol. All isoforms have deacetylase activity [82][83][84][85][86]. In fasting conditions, SIRT3 upregulated β-oxidation and ATP production [87], suppressed ROS, and increased mitochondrial biogenesis through activation of PGC1α [88]. Mice deficient in SIRT3 have hyperacetylated mitochondrial proteins [87]. SIRT3 is the highly expressed sirtuin in mouse liver [89]. SIRT3 has been shown to improve mitochondrial function and NAFLD by regulating β-oxidation, ketogenesis, mitophagy, and the antioxidant response system (Figure 2).
SIRT3 is downregulated in human and mouse models of NAFLD. SIRT3 and PGC1α can regulate each other, and both are reduced in HFD-fed mice [88]. Downregulation of SIRT3 with HFD feeding in mice induced hyperacetylation of mitochondrial proteins and increased hepatic fat storage and oxidative stress [88][90] (Figure 2). Exposure of mice lacking SIRT3 to HFD further increased the acetylation status of liver proteins and reduced respiratory complexes III and IV activity and increased oxidative stress [91][92]. Palmitate-induced lipotoxicity enhances ROS production and hepatocyte death in SIRT3-deficient primary hepatocytes [93][94]. SIRT3 overexpression reversed the suppression of ATP production induced by palmitate treatment [93][95]. In addition, SIRT3 overexpression repressed ROS generation [91]. HFD feeding in mice lacking SIRT3 exacerbated obesity, insulin resistance, hyper-lipidemia, hepatic steatosis, and inflammation [91][95]. Adenoviral overexpression of SIRT3 in these mice rescued the phenotype [91][92]. In addition to its effect on the mitochondria, SIRT3 deficiency in the liver aggravated hepatic steatosis in HFD-fed mice through upregulation of proteins involved in the FA uptake, such as CD36 and the VLDL receptor [94].
SIRT3 activates multiple targets such as long-chain acyl-CoA dehydrogenase (LCAD) and the acetyl-CoA synthase (AceCS) for acetyl-CoA formation [53]. RWesearchers have recently shown that SIRT3 is downregulated in the mitochondrial trifunctional protein heterozygous (MTP+/−) mice [89]. Overexpression of SIRT3 in MTP+/− mice deacetylates MTP, increases hepatic levels, and increases mitochondrial function [89]. SIRT3 is a positive regulator of autophagy and macroautophagy [96]. In primary hepatocytes from HFD-fed mice and mouse hepatocytes exposed to palmitic and oleic acid mixture, lipotoxicity decreased SIRT3 expression and lipophagic flux [96]. The decrease in lipophagy further worsened LDs accumulation, eventually leading to severe steatosis and hepatotoxicity. However, SIRT3 overexpression promoted macroautophagy in LDs through activating AMPK [96]. Treatment with Honokiol, a SIRT3 agonist, attenuated hepatic lipotoxicity by promoting SIRT3-AMPK-mediated lipophagy on lipid droplets [96]. In addition to lipophagy, SIRT3 has been shown to regulate mitophagy. Downregulation of SIRT3 with HFD-feeding inhibited Bnip3-mediated mitophagy, causing mitochondria-dependent hepatocyte death [95]. Furthermore, SIRT3 deletion aggravated hepatic steatosis, inflammation, and fibrogenesis in the methionine choline (MCD) mouse model of NAFLD partly by reducing the activity of the antioxidant enzyme SOD2 [97]. SIRT3 over-expression alleviated the MCD-induced phenotype [97], implicating SIRT3 deletion in NASH aggravation with MCD.
SIRTs are a potential therapeutic target for NAFLD as they protect hepatocytes against lipotoxicity [96]. The current state of sirtuin-targeted drug discovery and development has been recently reviewed in [98][99]. Small molecule sirtuin regulators have been developed, with a few compounds targeting human SIRTs still being in clinical development. The fundamental issues are the identification of isoform-specific and site-specific delivery of SIRTs activators [99][100].
References
- Hallsworth, K.; McPherson, S.; Anstee, Q.M.; Flynn, D.; Haigh, L.; Avery, L. Digital Intervention With Lifestyle Coach Support to Target Dietary and Physical Activity Behaviors of Adults With Nonalcoholic Fatty Liver Disease: Systematic Development Process of VITALISE Using Intervention Mapping. J. Med. Internet Res. 2021, 23, e20491.
- Sullivan, S.; Kirk, E.P.; Mittendorfer, B.; Patterson, B.W.; Klein, S. Randomized trial of exercise effect on intrahepatic triglyceride content and lipid kinetics in nonalcoholic fatty liver disease. Hepatology 2012, 55, 1738–1745.
- van der Windt, D.J.; Sud, V.; Zhang, H.; Tsung, A.; Huang, H. The Effects of Physical Exercise on Fatty Liver Disease. Gene Expr. 2018, 18, 89–101.
- Romero-Gomez, M.; Zelber-Sagi, S.; Trenell, M. Treatment of NAFLD with diet, physical activity and exercise. J. Hepatol. 2017, 67, 829–846.
- Vilar-Gomez, E.; Martinez-Perez, Y.; Calzadilla-Bertot, L.; Torres-Gonzalez, A.; Gra-Oramas, B.; Gonzalez-Fabian, L.; Friedman, S.L.; Diago, M.; Romero-Gomez, M. Weight Loss Through Lifestyle Modification Significantly Reduces Features of Nonalcoholic Steatohepatitis. Gastroenterology 2015, 149, 367–378.e365, quiz e314–365.
- Shojaee-Moradie, F.; Cuthbertson, D.; Barrett, M.; Jackson, N.; Herring, R.; Thomas, E.; Bell, J.; Kemp, G.; Wright, J.; Umpleby, A. Exercise training reduces liver fat and increases rates of VLDL clearance but not VLDL production in NAFLD. J. Clin. Endocrinol. Metab. 2016, 101, 4219–4228.
- Hallsworth, K.; Adams, L.A. Lifestyle modification in NAFLD/NASH: Facts and figures. JHEP Rep. Innov. Hepatol. 2019, 1, 468–479.
- Dulai, P.S.; Singh, S.; Patel, J.; Soni, M.; Prokop, L.J.; Younossi, Z.; Sebastiani, G.; Ekstedt, M.; Hagstrom, H.; Nasr, P.; et al. Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: Systematic review and meta-analysis. Hepatology 2017, 65, 1557–1565.
- Vilar-Gomez, E.; Calzadilla-Bertot, L.; Wai-Sun Wong, V.; Castellanos, M.; Aller-de la Fuente, R.; Metwally, M.; Eslam, M.; Gonzalez-Fabian, L.; Alvarez-Quiñones Sanz, M.; Conde-Martin, A.F.; et al. Fibrosis Severity as a Determinant of Cause-Specific Mortality in Patients With Advanced Nonalcoholic Fatty Liver Disease: A Multi-National Cohort Study. Gastroenterology 2018, 155, 443–457.
- Golabi, P.; Locklear, C.T.; Austin, P.; Afdhal, S.; Byrns, M.; Gerber, L.; Younossi, Z.M. Effectiveness of exercise in hepatic fat mobilization in non-alcoholic fatty liver disease: Systematic review. World J. Gastroenterol. 2016, 22, 6318–6327.
- El-Agroudy, N.N.; Kurzbach, A.; Rodionov, R.N.; O’Sullivan, J.; Roden, M.; Birkenfeld, A.L.; Pesta, D.H. Are Lifestyle Therapies Effective for NAFLD Treatment? Trends Endocrinol. Metab. 2019, 30, 701–709.
- Lazarus, J.V.; Mark, H.E.; Anstee, Q.M.; Arab, J.P.; Batterham, R.L.; Castera, L.; Cortez-Pinto, H.; Crespo, J.; Cusi, K.; Dirac, M.A.; et al. Advancing the global public health agenda for NAFLD: A consensus statement. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 60–78.
- Moore, M.P.; Cunningham, R.P.; Dashek, R.J.; Mucinski, J.M.; Rector, R.S. A Fad too Far? Dietary Strategies for the Prevention and Treatment of NAFLD. Obesity 2020, 28, 1843–1852.
- Banitalebi, E.; Faramarzi, M.; Nasiri, S.; Mardaniyan, M.; Rabiee, V. Effects of different exercise modalities on novel hepatic steatosis indices in overweight women with type 2 diabetes. Clin. Mol. Hepatol. 2019, 25, 294–304.
- Oh, S.; Tsujimoto, T.; Kim, B.; Uchida, F.; Suzuki, H.; Iizumi, S.; Isobe, T.; Sakae, T.; Tanaka, K.; Shoda, J. Weight-loss-independent benefits of exercise on liver steatosis and stiffness in Japanese men with NAFLD. JHEP Rep. 2021, 3, 100253.
- Johnson, N.A.; Sachinwalla, T.; Walton, D.W.; Smith, K.; Armstrong, A.; Thompson, M.W.; George, J. Aerobic exercise training reduces hepatic and visceral lipids in obese individuals without weight loss. Hepatology 2009, 50, 1105–1112.
- O’Gorman, P.; Naimimohasses, S.; Monaghan, A.; Kennedy, M.; Melo, A.M.; Ní Fhloinn, D.; Doherty, D.G.; Beddy, P.; Finn, S.P.; Moore, J.B.; et al. Improvement in histological endpoints of MAFLD following a 12-week aerobic exercise intervention. Aliment. Pharmacol. Ther. 2020, 52, 1387–1398.
- Rector, R.S.; Uptergrove, G.M.; Morris, E.M.; Borengasser, S.J.; Laughlin, M.H.; Booth, F.W.; Thyfault, J.P.; Ibdah, J.A. Daily exercise vs. caloric restriction for prevention of nonalcoholic fatty liver disease in the OLETF rat model. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G874–G883.
- Thyfault, J.P.; Rector, R.S. Exercise Combats Hepatic Steatosis: Potential Mechanisms and Clinical Implications. Diabetes 2020, 69, 517–524.
- Farzanegi, P.; Dana, A.; Ebrahimpoor, Z.; Asadi, M.; Azarbayjani, M.A. Mechanisms of beneficial effects of exercise training on non-alcoholic fatty liver disease (NAFLD): Roles of oxidative stress and inflammation. Eur. J. Sport Sci. 2019, 19, 994–1003.
- Rector, R.S.; Thyfault, J.P.; Morris, R.T.; Laye, M.J.; Borengasser, S.J.; Booth, F.W.; Ibdah, J.A. Daily exercise increases hepatic fatty acid oxidation and prevents steatosis in Otsuka Long-Evans Tokushima Fatty rats. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G619–G626.
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357.
- Diehl, A.M.; Day, C. Cause, pathogenesis, and treatment of nonalcoholic steatohepatitis. N. Engl. J. Med. 2017, 377, 2063–2072.
- Lassailly, G.; Caiazzo, R.; Ntandja-Wandji, L.-C.; Gnemmi, V.; Baud, G.; Verkindt, H.; Ningarhari, M.; Louvet, A.; Leteurtre, E.; Raverdy, V. Bariatric surgery provides long-term resolution of nonalcoholic steatohepatitis and regression of fibrosis. Gastroenterology 2020, 159, 1290–1301.e1295.
- Fakhry, T.K.; Mhaskar, R.; Schwitalla, T.; Muradova, E.; Gonzalvo, J.P.; Murr, M.M. Bariatric surgery improves nonalcoholic fatty liver disease: A contemporary systematic review and meta-analysis. Surg Obes. Relat Dis 2019, 15, 502–511.
- Alkhouri, N.; Tincopa, M.; Loomba, R.; Harrison, S.A. What Does the Future Hold for Patients With Nonalcoholic Steatohepatitis: Diagnostic Strategies and Treatment Options in 2021 and Beyond? Hepatol. Commun. 2021, 5, 1810–1823.
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685.
- Lavine, J.E.; Schwimmer, J.B.; Van Natta, M.L.; Molleston, J.P.; Murray, K.F.; Rosenthal, P.; Abrams, S.H.; Scheimann, A.O.; Sanyal, A.J.; Chalasani, N.; et al. Effect of vitamin E or metformin for treatment of nonalcoholic fatty liver disease in children and adolescents: The TONIC randomized controlled trial. Jama 2011, 305, 1659–1668.
- Cusi, K.; Orsak, B.; Bril, F.; Lomonaco, R.; Hecht, J.; Ortiz-Lopez, C.; Tio, F.; Hardies, J.; Darland, C.; Musi, N.; et al. Long-Term Pioglitazone Treatment for Patients With Nonalcoholic Steatohepatitis and Prediabetes or Type 2 Diabetes Mellitus: A Randomized Trial. Ann. Intern. Med. 2016, 165, 305–315.
- Lange, N.F.; Graf, V.; Caussy, C.; Dufour, J.F. PPAR-Targeted Therapies in the Treatment of Non-Alcoholic Fatty Liver Disease in Diabetic Patients. Int. J. Mol. Sci. 2022, 23, 4305.
- Jiang, L.; Zhang, H.; Xiao, D.; Wei, H.; Chen, Y. Farnesoid X receptor (FXR): Structures and ligands. Comput. Struct. Biotechnol. J. 2021, 19, 2148–2159.
- Stofan, M.; Guo, G.L. Bile Acids and FXR: Novel Targets for Liver Diseases. Front. Med. 2020, 7, 544.
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; Van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 2015, 385, 956–965.
- Younossi, Z.M.; Ratziu, V.; Loomba, R.; Rinella, M.; Anstee, Q.M.; Goodman, Z.; Bedossa, P.; Geier, A.; Beckebaum, S.; Newsome, P.N.; et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: Interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2019, 394, 2184–2196.
- Sinha, R.A.; Singh, B.K.; Yen, P.M. Direct effects of thyroid hormones on hepatic lipid metabolism. Nat. Rev. Endocrinol. 2018, 14, 259–269.
- Harrison, S.A.; Bashir, M.; Moussa, S.E.; McCarty, K.; Pablo Frias, J.; Taub, R.; Alkhouri, N. Effects of Resmetirom on Noninvasive Endpoints in a 36-Week Phase 2 Active Treatment Extension Study in Patients With NASH. Hepatol. Commun. 2021, 5, 573–588.
- Iruarrizaga-Lejarreta, M.; Varela-Rey, M.; Fernández-Ramos, D.; Martínez-Arranz, I.; Delgado, T.C.; Simon, J.; Juan, V.G.; delaCruz-Villar, L.; Azkargorta, M.; Lavin, J.L.; et al. Role of Aramchol in steatohepatitis and fibrosis in mice. Hepatol. Commun. 2017, 1, 911–927.
- Ratziu, V.; de Guevara, L.; Safadi, R.; Poordad, F.; Fuster, F.; Flores-Figueroa, J.; Arrese, M.; Fracanzani, A.L.; Ben Bashat, D.; Lackner, K.; et al. Aramchol in patients with nonalcoholic steatohepatitis: A randomized, double-blind, placebo-controlled phase 2b trial. Nat. Med. 2021, 27, 1825–1835.
- Seino, Y.; Fukushima, M.; Yabe, D. GIP and GLP-1, the two incretin hormones: Similarities and differences. J. Diabetes Investig 2010, 1, 8–23.
- Newsome, P.N.; Buchholtz, K.; Cusi, K.; Linder, M.; Okanoue, T.; Ratziu, V.; Sanyal, A.J.; Sejling, A.S.; Harrison, S.A. A Placebo-Controlled Trial of Subcutaneous Semaglutide in Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2021, 384, 1113–1124.
- Yu, X.; Hao, M.; Liu, Y.; Ma, X.; Lin, W.; Xu, Q.; Zhou, H.; Shao, N.; Kuang, H. Liraglutide ameliorates non-alcoholic steatohepatitis by inhibiting NLRP3 inflammasome and pyroptosis activation via mitophagy. Eur. J. Pharmacol. 2019, 864, 172715.
- Vallon, V.; Verma, S. Effects of SGLT2 Inhibitors on Kidney and Cardiovascular Function. Annu. Rev. Physiol. 2021, 83, 503–528.
- Shimizu, M.; Suzuki, K.; Kato, K.; Jojima, T.; Iijima, T.; Murohisa, T.; Iijima, M.; Takekawa, H.; Usui, I.; Hiraishi, H.; et al. Evaluation of the effects of dapagliflozin, a sodium-glucose co-transporter-2 inhibitor, on hepatic steatosis and fibrosis using transient elastography in patients with type 2 diabetes and non-alcoholic fatty liver disease. Diabetes Obes. Metab. 2019, 21, 285–292.
- Henriksson, E.; Andersen, B. FGF19 and FGF21 for the Treatment of NASH-Two Sides of the Same Coin? Differential and Overlapping Effects of FGF19 and FGF21 From Mice to Human. Front. Endocrinol. 2020, 11, 601349.
- Talukdar, S.; Kharitonenkov, A. FGF19 and FGF21: In NASH we trust. Mol. Metab. 2021, 46, 101152.
- Harrison, S.A.; Ruane, P.J.; Freilich, B.L.; Neff, G.; Patil, R.; Behling, C.A.; Hu, C.; Fong, E.; de Temple, B.; Tillman, E.J.; et al. Efruxifermin in non-alcoholic steatohepatitis: A randomized, double-blind, placebo-controlled, phase 2a trial. Nat. Med. 2021, 27, 1262–1271.
- Bartesaghi, S.; Wallenius, K.; Hovdal, D.; Liljeblad, M.; Wallin, S.; Dekker, N.; Barlind, L.; Davies, N.; Seeliger, F.; Winzell, M.S.; et al. Subcutaneous delivery of FGF21 mRNA therapy reverses obesity, insulin resistance, and hepatic steatosis in diet-induced obese mice. Mol. Ther. Nucleic Acids 2022, 28, 500–513.
- Ciardullo, S.; Perseghin, G. Statin use is associated with lower prevalence of advanced liver fibrosis in patients with type 2 diabetes. Metabolism 2021, 121, 154752.
- Ding, R.-B.; Bao, J.; Deng, C.-X. Emerging roles of SIRT1 in fatty liver diseases. Int. J. Biol. Sci. 2017, 13, 852–867.
- Nassir, F. Role of acetylation in nonalcoholic fatty liver disease: A focus on SIRT1 and SIRT3. Explor. Med. 2020, 1, 248–258.
- Nassir, F.; Ibdah, J.A. Sirtuins and nonalcoholic fatty liver disease. World J. Gastroenterol. 2016, 22, 10084–10092.
- Hirschey, M.D.; Shimazu, T.; Goetzman, E.; Jing, E.; Schwer, B.; Lombard, D.B.; Grueter, C.A.; Harris, C.; Biddinger, S.; Ilkayeva, O.R. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 2010, 464, 121–125.
- Kulkarni, S.S.; Cantó, C. Mitochondrial Post-translational Modifications and Metabolic Control: Sirtuins and Beyond. Curr. Diabetes Rev. 2017, 13, 338–351.
- Nogueiras, R.; Habegger, K.M.; Chaudhary, N.; Finan, B.; Banks, A.S.; Dietrich, M.O.; Horvath, T.L.; Sinclair, D.A.; Pfluger, P.T.; Tschöp, M.H. Sirtuin 1 and sirtuin 3: Physiological modulators of metabolism. Physiol. Rev. 2012, 92, 1479–1514.
- Singh, C.K.; Chhabra, G.; Ndiaye, M.A.; Garcia-Peterson, L.M.; Mack, N.J.; Ahmad, N. The Role of Sirtuins in Antioxidant and Redox Signaling. Antioxid. Redox Signal. 2018, 28, 643–661.
- Tanno, M.; Sakamoto, J.; Miura, T.; Shimamoto, K.; Horio, Y. Nucleocytoplasmic shuttling of the NAD+-dependent histone deacetylase SIRT1. J. Biol. Chem. 2007, 282, 6823–6832.
- Lee, I.H.; Cao, L.; Mostoslavsky, R.; Lombard, D.B.; Liu, J.; Bruns, N.E.; Tsokos, M.; Alt, F.W.; Finkel, T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. USA 2008, 105, 3374–3379.
- Majeed, Y.; Halabi, N.; Madani, A.Y.; Engelke, R.; Bhagwat, A.M.; Abdesselem, H.; Agha, M.V.; Vakayil, M.; Courjaret, R.; Goswami, N.; et al. SIRT1 promotes lipid metabolism and mitochondrial biogenesis in adipocytes and coordinates adipogenesis by targeting key enzymatic pathways. Sci. Rep. 2021, 11, 8177.
- Wu, T.; Liu, Y.H.; Fu, Y.C.; Liu, X.M.; Zhou, X.H. Direct evidence of sirtuin downregulation in the liver of non-alcoholic fatty liver disease patients. Ann. Clin. Lab. Sci. 2014, 44, 410–418.
- Cheng, J.; Liu, C.; Hu, K.; Greenberg, A.; Wu, D.; Ausman, L.M.; McBurney, M.W.; Wang, X.-D. Ablation of systemic SIRT1 activity promotes nonalcoholic fatty liver disease by affecting liver-mesenteric adipose tissue fatty acid mobilization. Biochim. Et Biophys. Acta BBA Mol. Basis Dis. 2017, 1863, 2783–2790.
- Mariani, S.; Fiore, D.; Basciani, S.; Persichetti, A.; Contini, S.; Lubrano, C.; Salvatori, L.; Lenzi, A.; Gnessi, L. Plasma levels of SIRT1 associate with non-alcoholic fatty liver disease in obese patients. Endocrine 2014, 49, 711–716.
- Liang, G.; Yang, J.; Horton, J.D.; Hammer, R.E.; Goldstein, J.L.; Brown, M.S. Diminished hepatic response to fasting/refeeding and liver X receptor agonists in mice with selective deficiency of sterol regulatory element-binding protein-1c. J. Biol. Chem. 2002, 277, 9520–9528.
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 2005, 434, 113–118.
- Khan, S.A.; Sathyanarayan, A.; Mashek, M.T.; Ong, K.T.; Wollaston-Hayden, E.E.; Mashek, D.G. ATGL-catalyzed lipolysis regulates SIRT1 to control PGC-1α/PPAR-α signaling. Diabetes 2015, 64, 418–426.
- Purushotham, A.; Schug, T.T.; Xu, Q.; Surapureddi, S.; Guo, X.; Li, X. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 2009, 9, 327–338.
- Bordone, L.; Cohen, D.; Robinson, A.; Motta, M.C.; van Veen, E.; Czopik, A.; Steele, A.D.; Crowe, H.; Marmor, S.; Luo, J.; et al. SIRT1 transgenic mice show phenotypes resembling calorie restriction. Aging Cell 2007, 6, 759–767.
- Wang, R.-H.; Li, C.; Deng, C.-X. Liver steatosis and increased ChREBP expression in mice carrying a liver specific SIRT1 null mutation under a normal feeding condition. Int. J. Biol. Sci. 2010, 6, 682.
- Ren, T.; Zhu, L.; Shen, Y.; Mou, Q.; Lin, T.; Feng, H. Protection of hepatocyte mitochondrial function by blueberry juice and probiotics via SIRT1 regulation in non-alcoholic fatty liver disease. Food Funct. 2019, 10, 1540–1551.
- Cantó, C.; Auwerx, J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 2009, 20, 98–105.
- Gureev, A.P.; Shaforostova, E.A.; Popov, V.N. Regulation of Mitochondrial Biogenesis as a Way for Active Longevity: Interaction Between the Nrf2 and PGC-1α Signaling Pathways. Front. Genet. 2019, 10.
- Towler, M.C.; Hardie, D.G. AMP-activated protein kinase in metabolic control and insulin signaling. Circ. Res. 2007, 100, 328–341.
- Cantó, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060.
- Jäger, S.; Handschin, C.; St-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022.
- de Gregorio, E.; Colell, A.; Morales, A.; Marí, M. Relevance of SIRT1-NF-κB Axis as Therapeutic Target to Ameliorate Inflammation in Liver Disease. Int. J. Mol. Sci. 2020, 21, 3858.
- Price, N.L.; Gomes, A.P.; Ling, A.J.Y.; Duarte, F.V.; Martin-Montalvo, A.; North, B.J.; Agarwal, B.; Ye, L.; Ramadori, G.; Teodoro, J.S.; et al. SIRT1 Is Required for AMPK Activation and the Beneficial Effects of Resveratrol on Mitochondrial Function. Cell Metab. 2012, 15, 675–690.
- Xiong, X.; Yu, J.; Fan, R.; Zhang, C.; Xu, L.; Sun, X.; Huang, Y.; Wang, Q.; Ruan, H.B.; Qian, X. NAMPT overexpression alleviates alcohol-induced hepatic steatosis in mice. PLoS ONE 2019, 14, e0212523.
- Audrito, V.; Messana, V.G.; Deaglio, S. NAMPT and NAPRT: Two Metabolic Enzymes With Key Roles in Inflammation. Front. Oncol 2020, 10, 358.
- Dall, M.; Hassing, A.S.; Treebak, J.T. NAD(+) and NAFLD–caution, causality and careful optimism. J. Physiol. 2022, 600, 1135–1154.
- Imai, S.; Yoshino, J. The importance of NAMPT/NAD/SIRT1 in the systemic regulation of metabolism and ageing. Diabetes Obes. Metab. 2013, 15 (Suppl. 3), 26–33.
- Wang, Y.; Zhu, K.; Yu, W.; Wang, H.; Liu, L.; Wu, Q.; Li, S.; Guo, J. MiR-181b regulates steatosis in nonalcoholic fatty liver disease via targeting SIRT1. Biochem. Biophys. Res. Commun. 2017, 493, 227–232.
- Zhang, J.; Xiang, H.; Liu, J.; Chen, Y.; He, R.R.; Liu, B. Mitochondrial Sirtuin 3: New emerging biological function and therapeutic target. Theranostics 2020, 10, 8315–8342.
- Jin, L.; Galonek, H.; Israelian, K.; Choy, W.; Morrison, M.; Xia, Y.; Wang, X.; Xu, Y.; Yang, Y.; Smith, J.J. Biochemical characterization, localization, and tissue distribution of the longer form of mouse SIRT3. Protein Sci. 2009, 18, 514–525.
- Yang, Y.; Hubbard, B.P.; Sinclair, D.A.; Tong, Q. Characterization of murine SIRT3 transcript variants and corresponding protein products. J. Cell. Biochem. 2010, 111, 1051–1058.
- Yang, Y.; Chen, K.Y.; Tong, Q. Murine Sirt3 protein isoforms have variable half-lives. Gene 2011, 488, 46–51.
- Lombard, D.B.; Alt, F.W.; Cheng, H.-L.; Bunkenborg, J.; Streeper, R.S.; Mostoslavsky, R.; Kim, J.; Yancopoulos, G.; Valenzuela, D.; Murphy, A. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol. Cell. Biol. 2007, 27, 8807–8814.
- Schwer, B.; Bunkenborg, J.; Verdin, R.O.; Andersen, J.S.; Verdin, E. Reversible lysine acetylation controls the activity of the mitochondrial enzyme acetyl-CoA synthetase 2. Proc. Natl. Acad. Sci. USA 2006, 103, 10224–10229.
- Ahn, B.H.; Kim, H.S.; Song, S.; Lee, I.H.; Liu, J.; Vassilopoulos, A.; Deng, C.X.; Finkel, T. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc. Natl. Acad. Sci. USA 2008, 105, 14447–14452.
- Kong, X.; Wang, R.; Xue, Y.; Liu, X.; Zhang, H.; Chen, Y.; Fang, F.; Chang, Y. Sirtuin 3, a new target of PGC-1α, plays an important role in the suppression of ROS and mitochondrial biogenesis. PLoS ONE 2010, 5, e11707.
- Nassir, F.; Arndt, J.J.; Johnson, S.A.; Ibdah, J.A. Regulation of mitochondrial trifunctional protein modulates nonalcoholic fatty liver disease in mice. J. Lipid Res. 2018, 59, 967–973.
- Bao, J.; Lu, Z.; Joseph, J.J.; Carabenciov, D.; Dimond, C.C.; Pang, L.; Samsel, L.; McCoy, J.P., Jr.; Leclerc, J.; Nguyen, P.; et al. Characterization of the murine SIRT3 mitochondrial localization sequence and comparison of mitochondrial enrichment and deacetylase activity of long and short SIRT3 isoforms. J. Cell. Biochem. 2010, 110, 238–247.
- Hirschey, M.D.; Shimazu, T.; Jing, E.; Grueter, C.A.; Collins, A.M.; Aouizerat, B.; Stancakova, A.; Goetzman, E.; Lam, M.M.; Schwer, B.; et al. SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol. Cell 2011, 44, 177–190.
- Kendrick, A.A.; Choudhury, M.; Rahman, S.M.; McCurdy, C.E.; Friederich, M.; Van Hove, J.L.; Watson, P.A.; Birdsey, N.; Bao, J.; Gius, D. Fatty liver is associated with reduced SIRT3 activity and mitochondrial protein hyperacetylation. Biochem. J. 2011, 433, 505–514.
- Bao, J.; Scott, I.; Lu, Z.; Pang, L.; Dimond, C.C.; Gius, D.; Sack, M.N. SIRT3 is regulated by nutrient excess and modulates hepatic susceptibility to lipotoxicity. Free Radic. Biol. Med. 2010, 49, 1230–1237.
- Barroso, E.; Rodríguez-Rodríguez, R.; Zarei, M.; Pizarro-Degado, J.; Planavila, A.; Palomer, X.; Villarroya, F.; Vázquez-Carrera, M. SIRT3 deficiency exacerbates fatty liver by attenuating the HIF1α-LIPIN 1 pathway and increasing CD36 through Nrf2. Cell Commun. Signal. 2020, 18, 147.
- Li, R.; Xin, T.; Li, D.; Wang, C.; Zhu, H.; Zhou, H. Therapeutic effect of Sirtuin 3 on ameliorating nonalcoholic fatty liver disease: The role of the ERK-CREB pathway and Bnip3-mediated mitophagy. Redox Biol. 2018, 18, 229–243.
- Zhang, T.; Liu, J.; Shen, S.; Tong, Q.; Ma, X.; Lin, L. SIRT3 promotes lipophagy and chaperon-mediated autophagy to protect hepatocytes against lipotoxicity. Cell Death Differ. 2020, 27, 329–344.
- He, J.; Hu, B.; Shi, X.; Weidert, E.; Lu, P.; Xu, M.; Huang, M.; Kelley, E.; Xie, W. Activation of the Aryl Hydrocarbon Receptor Sensitizes Mice to Nonalcoholic Steatohepatitis by Deactivating Mitochondrial Sirtuin Deacetylase Sirt3. Mol. Cell. Biol. 2013, 33.
- Martinou, E.; Pericleous, M.; Stefanova, I.; Kaur, V.; Angelidi, A.M. Diagnostic Modalities of Non-Alcoholic Fatty Liver Disease: From Biochemical Biomarkers to Multi-Omics Non-Invasive Approaches. Diagnostics 2022, 12, 407.
- Dai, H.; Sinclair, D.A.; Ellis, J.L.; Steegborn, C. Sirtuin activators and inhibitors: Promises, achievements, and challenges. Pharmacol. Ther. 2018, 188, 140–154.
- Curry, A.M.; White, D.S.; Donu, D.; Cen, Y. Human Sirtuin Regulators: The "Success" Stories. Front. Physiol. 2021, 12, 752117.
More