Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Sasinee Hantrakool and Version 2 by Sirius Huang.
Ambient air pollution has become a common problem worldwide. Exposure to pollutant particles causes many health conditions, having a particular impact on pulmonary and cardiovascular disease. Increased understanding of the pathological processes related to these conditions may facilitate the prevention of the adverse impact of air pollution on our physical health.
- particulate matter
- inflammation
- oxidative stress
- adhesion molecule
- coagulation
- thrombosis
1. Introduction
Air pollution has recently become a major concern worldwide. It has been proposed that exposure to high ambient pollutant particles leads to adverse health impacts which may contribute to as many as three million premature deaths per year [1][2][1,2]. There is extensive evidence to confirm the various adverse outcomes of the small particles on our health after both acute and long-term exposure [3]. These fine particles enter the blood circulation through the respiratory tract and quickly activate the pulmonary and systemic inflammatory responses. Activation of the inflammatory cytokines, oxidative stress, and adhesion molecules results in atherosclerosis [4][5][6][4,5,6]. Previous reports revealed a significant association between high ambient particulate matter and an increased incidence of cardiovascular disease including ischemic stroke [7][8][7,8], acute coronary syndrome [9][10][11][9,10,11], and thrombosis [12][13][14][12,13,14].
Particulate matter (PM) can consist of a variety of components depending on the source [15][16][15,16]. PM is defined according to particle size. Coarse particulate matter, PM10 has an aerodynamic diameter of between 2.5 to 10 microns. Fine particulate matter, PM2.5 has a particle size of more than 0.1 micron, but less than 2.5 microns in aerodynamic diameter. Ultrafine particulate matter (UFP) has an aerodynamic diameter of less than 0.1 micron [17][18][17,18]. Diesel exhaust particles (DEP) are the particles released from the combustion of fuel in diesel-fuelled vehicles and consist of a mixture of polycyclic aromatic hydrocarbon, organic compounds, sulfate, nitrate, other trace elements, and metals such as copper, iron, nickel, vanadium, and zinc [19]. These particles enter the body via inhalation. The coarse particles are mostly trapped in the upper airway, whereas smaller particles can pass beyond the lower airway, passing into the blood, causing adverse effects throughout the body [17][18][17,18]. Past evidence has demonstrated that PM could induce oxidative stress [20][21][22][23][24][25][26][27][20,21,22,23,24,25,26,27], result in DNA damage [25][28][29][25,28,29], and activate local and systemic inflammatory response [22][23][25][26][30][22,23,25,26,30]. Furthermore, PM has also been shown to impair vascular function [30][31][30,31], increase the expression of vascular inflammatory biomarkers, including intercellular adhesion molecules (ICAM-1), vascular cell adhesion molecules (VCAM-1), and P-selectin [24][32][24,32]. VCAM-1 and ICAM-1 and p-selectin are vascular adhesion molecules that play an important role in thrombus formation by promoting leukocyte-endothelial and leukocyte-platelet interaction during the inflammatory response [24][32][24,32]. Increased levels of these adhesion molecules instigate the recruitment of inflammatory cells into vascular endothelium resulting in the release of microparticles and the activation of platelet adhesion and platelet aggregation. Blood microparticles are small parts of cell membrane, secreted from various cell types, including endothelial cells, monocytes, and platelets, and containing both phosphatidylserine and tissue factor, the potent procoagulants, which further activate the down-stream coagulation cascade [33][34][33,34].
2. Hemostasis and the Fibrinolytic Pathway
Normal hemostasis is a complex system mainly maintaining stable physiology in the body and protecting against pathological processes. The generation of thrombin is the key mechanism that links blood clot formation and the fibrinolytic system, the counterbalance that controls the thrombotic process [33][35][33,35]. In general, vascular endothelial injury induces platelet adhesion, platelet activation, and the release of the von Willebrand factor (vWF), tissue factor (TF), cytokines and chemokines. Primary hemostasis occurs when platelets adhere to the injured site, activating platelet aggregation, which is promoted by the vWF. TF is the main coagulation factor that initiates blood coagulation by activation of Factor VII in the extrinsic pathway, resulting in the generation of thrombin. This would further stimulate elements of the intrinsic pathway including Factors IX and VIII and then the downstream common coagulation pathway including Factors X and V, resulting in production of the prothrombinase complex (FVa-Xa), which generates excessive thrombin, leading to fibrin clot formation [33][35][33,35]. In the fibrinolytic pathway, tissue plasminogen activator (tPA) and plasminogen-activator inhibitor-1 (PAI-1) are the main regulators that modulate fibrinolysis. tPA activates and causes the cleavage of plasminogen into plasmin, resulting in the degradation of fibrin clots. PAI-1 is a serine protease inhibitor, which inhibits tPA, preventing clot lysis. An imbalance of tPA and PAI-1 levels therefore has an impact on fibrin clot formation [35]. Hemostasis and fibrinolytic pathways are summarized in Figure 1.
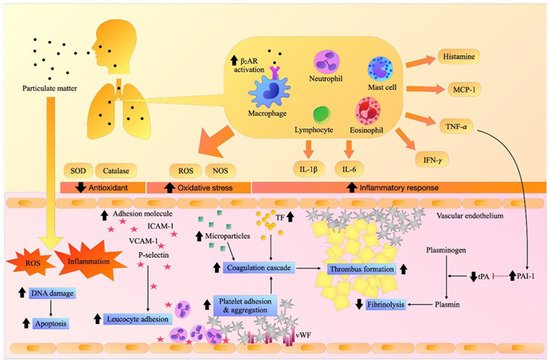
Figure 1.
Effects of particulate matter (PM) on inflammation, oxidative stress, adhesion molecules, and thrombosis.
Inhalation exposure to PM induces pulmonary and systemic inflammation and oxidative stress. It increases the expression of adhesion molecules resulting in the recruitment of inflammatory cells and the activation of the vascular endothelium, platelets, and coagulation cascade, causing fibrin clots, while hampering fibrinolytic activity, and eventually contributing to thrombosis. β2AR: beta-2 adrenergic receptor, DNA: deoxyribonucleic acid, ICAM-1: intercellular adhesion molecules-1, IFN-γ. interferon- γ, IL-1β: interleukin-1β, IL-6: interleukin-6, MCP-1: monocyte chemoattractant protein-1, NOS: nitric oxide synthase, PAI-1: plasminogen activator inhibitor-1, ROS: reactive oxygen species, SOD: superoxide dismutase, TF: tissue factor, TNF-α: tumor necrosis factor-α, tPA: tissue plasminogen activator, VCAM-1: vascular adhesion molecule-1.
Currently, knowledge surrounding the outcomes of PM-mediated coagulation and fibrinolysis is still inconclusive. In theis next sectionsreview, the effects of PM on the inflammatory responses, oxidative stress, adhesion molecules, and coagulation factors related to thrombosis from in vitro, in vivo, and clinical studies are comprehensively summarized and discussed. The understanding accrued herefrom this overview of the pathological process of PM-mediated thrombosis will help in limiting or preventing the damaging effects of PM exposure on our health.
3. The Effects of Particulate Matter on Inflammation, Oxidative Stress, and the Coagulation System: Reports from In Vitro Studies
Over the past decade, it has been shown that polluted air particles can activate inflammation, and oxidative stress and cause cell death [36][37][36,37]. In vitro reports indicate that exposure to PM could enhance the inflammatory response and oxidative stress, activating the coagulation cascade and inducing cell death, leading to a prothrombotic state [38]. Vanadium pentoxide (V2O5) is one of the toxic substances, a consequence of burning fuel oil and fly ash. An in vitro study showed that V2O5 could directly affect the human umbilical vein endothelial cells (HUVECs), by enhancing oxidative stress, and increasing expression of adhesion molecules, which resulted in shape changes, decreased cell proliferation, and increased apoptosis [38]. A report revealed that murine peritoneal macrophages incubated with urban PM could induce inflammatory cytokines release, resulting in phenotype changes (M1/M2 polarization), cell injury, and decreased engulfment function [39]. Other reports had shown that PM exposure induced lung macrophage differentiation into a more pro-inflammatory subtype (M1-phenotype) rather than an anti-inflammatory subpopulation (M2-phenotype) [40][41][40,41], which attenuates the phagocytic activity against bacterial invasion, and probably led in more susceptible to pulmonary infection [39][40][41][42][43][39,40,41,42,43].
PM could also induce cAMP secretion and activate further downstream pathways, resulting in the enhancement of PM-mediated IL-6 release in murine alveolar macrophages (MH-S) and human alveolar macrophages [44]. In addition, administration of albuterol, a β2AR agonist, enhanced PM-induced IL-6 release in human alveolar macrophages and MH-S cell lines, while the alveolar macrophages from Adrb2−/− mice incubated with albuterol showed a decreased PM-mediated IL-6 response [44]. These findings suggest that PM-mediated IL-6 release was dependent on the activation of β2AR, encoded by the Adrb2 gene. Moreover, PM was also shown to activate the microparticles and cause intracellular calcium release, and to enhance tissue factor function in HUVECs and peripheral blood mononuclear cells (PBMCs) [45]. The activation of tissue factors would further trigger blood coagulation via the TF-FVII complex, leading to thrombus formation.
In in vitro studies of venous blood from rodent models, incubation with DEP rapidly induced platelet activation and platelet aggregation in a dose-dependent manner [46][47][48][49][46,47,48,49]. This effect was emphasized in diabetic mice, which were more vulnerable to thrombotic complications [49]. In addition, incubation of HUVECs with DEP revealed decreasing tPA and PAI-1 activity [50]. These findings suggested that both DEP and PM induced thrombosis by activating the tissue factor pathway and enhancing platelet aggregation, as well as inhibiting the fibrinolytic process, thus promoting blood clot formation and thrombosis.
The different types of particles also had differing effects on outcomes. Positively-charged amine-particles could enhance platelet function, as indicated by the shortening of PFA100 closure time in both 60-nm UFP and 400-nm amine-polystyrene particles. In contrast, negatively-charged carboxylated UFP and unmodified UFP did not affect platelet function [51]. This means that the character of each particle also plays a role in PM-induced platelet aggregation. A summary of these in vitro reports on the effects of PM on inflammation, oxidative stress, adhesion molecules, coagulation, and cell proliferation is shown in Table 1.
Table 1. The effects of particulate matter on inflammation, oxidative stress, adhesion molecules, and hemostatic changes: Evidence from in vitro studies.
| Models | Exposure/Method | Results | Interpretation | References | ||
|---|---|---|---|---|---|---|
| Inflammation and Oxidative Stress | Coagulation and Adhesion Molecules | Morphology and Cell Proliferation | ||||
| HUVECs | Vanadium oxide (V2O5) 3.12, 6.25, 12.5, 25 µg/cm2 for 1, 2, 3, 24, 48, 72 h |
↑ ROS at 25 µg/cm2 ↑ NO at 25 µg/cm2 (time-dependent) |
↑ VCAM-1 ↑ ICAM-1 ↑↑ PECAM-1 |
Morphology changed to fibroblast-like cells ↓ cell proliferation at 25 µg/cm2 ↑ annexin V, PI |
Exposure to V2O5 induced oxidative stress, enhanced the expression of adhesion molecules, and affected cell survival by diminishing cell proliferation, shape changes, and apoptosis. | [38] |
| MH-S, Human alveolar macrophages |
PM (SRM 1649a) 10 µg/cm2 for 24 h |
# MH-S: ↑ IL-6 ↑ cAMP # Human alveolar macrophage: ↑ IL-6 |
β2AR encoding from the Adrb2 gene had an important role in PM-induced IL-6 release and activation of β2AR enhanced inflammatory response in both cell lines. | [44] | ||
| PM (SRM 1649a) 10 µg/cm2 for 24 h Pretreated with β2AR agonist; albuterol 10−7 M |
↑↑ IL-6 | |||||
| Alveolar macrophages from Adrb2−/− mice | PM (SRM 1649a) 10 µg/cm2 for 24 h Pretreated with β2AR agonist; albuterol 10−7 M |
↓ IL-6 | ||||
| HUVECs, PBMC | PM (SRM 1648a) 62.5, 125, 250 and 500 µg/mL for 1, 4, 24, 48 h |
↑ MP (dose-, and time-dependent) ↑ intracellular Ca concentration |
↑ TF activity | PM-induced MP release, which, mediated by calcium mobilization, resulted in the prothrombotic state in both cell lines. | [45] | |
| HUVECs | DEP 10–150 µg/mL for 16 h ±thrombin stimulation 1 U/mL |
# Without thrombin: ↓ tPA ↓ PAI-1 # With thrombin stimulation: ↔ tPA ↑ PAI-1 |
DEP enhanced arterial thrombus formation through decreased fibrinolytic function but did not affect cell survival. | [50] | ||
| Venous blood of hamsters |
DEP (SRM 1650) 0.1, 0.5, 1, 5 µg/mL for 5 min |
↓ PFA100 closure time, dose-dependent |
DEP promoted thrombosis via platelet activation in a dose-dependent manner. | [46] | ||
| Venous blood of TO mice |
DEP 1 µg/mL for 3 min |
↑ platelet aggregation ↓ PT ↓ PTT |
DEP promoted thrombosis by enhancing platelet aggregation and coagulation. | [47] | ||
| Venous blood of TO mice |
DEP (SRM 2975) 0.1, 0.25, 0.5, 1 µg/mL for 3 min |
↑ platelet aggregation at 0.5 and 1 µg/mL, dose-dependent | DEP promoted thrombosis by enhancing platelet aggregation. | [48] | ||
| Venous blood of TO mice Non-DM and DM mice |
DEP (SRM 2975) 0.25, 0.5, 1 µg/mL for 3 min |
# Non-DM mice: ↑ platelet aggregation at 1 µg/mL # DM mice: ↑↑ platelet aggregation, dose-dependent |
DEP promoted thrombosis by enhancing platelet aggregation, which was more obvious in DM mice. | [49] | ||
| Venous blood of hamsters (Pfd Gold) |
Polystyrene particles: # 60 nm UFP - unmodified - carboxylated - amined at 1 or 3 µg/mL # 400 nm: Amine-polystyrene particles at 3 or 9 µg/mL for 5 min |
# Unmodified and carboxylated UFP: ↔ PFA100 closure time # Amine-UFP (60 nm): ↓ PFA100 closure time (3 µg/mL) # Amine-particles (400 nm): ↓ PFA100 closure time (9 µg/mL) |
Exposure to positively charged UFP (60 & 400 nm) augmented platelet function, leading to thrombosis. | [51] | ||
Ca: calcium, cAMP: cyclic adenosine monophosphate, DEP: diesel exhaust particles, DM: diabetes mellitus, HUVECs: human umbilical vein endothelial cells, h: hours, ICAM-1: intercellular adhesion molecule-1, IL-6: interleukin-6, MH-S: murine alveolar macrophage cell line, min: minutes, MP: microparticles, NO: nitric oxide, PAI-1: plasminogen activator inhibitor-1, PBMC: peripheral blood mononuclear cells, PECAM-1: platelet endothelial cell adhesion molecule-1, PFA100: platelet function analyzer-100, PI: propidium iodide, PM: particulate matter, PT: prothrombin time, PTT: partial thromboplastin time, ROS: reactive oxygen species, SRM: standard reference material, TF: tissue factor, TO mice: Tuck-Ordinary mice, tPA: tissue plasminogen activator, UFP: ultrafine particles, VCAM-1: vascular cell adhesion molecule-1, V2O5: Vanadium oxide.
4. The Effects of Particulate Matter on Inflammation, Oxidative Stress, and the Coagulation System: Reports from In Vivo Studies
Consistent with in vitro reports, in vivo studies showed that the β2-adrenergic receptor (β2AR) in the macrophages encoded for by the Adrb2 gene was the key receptor that modulates PM-induced inflammation and thrombosis [44]. Adrb2-knockout mice had increased the PM-mediated IL-6 release, whereas this effect was blunted in specific Lyms-Cre Adrb2flox/flox mice together with a decrease in thrombus formation and tissue factors [44]. In addition, Lyms-Cre Adrb2flox/flox mice pretreated with a β2AR agonist, Formoterol, showed that these mice which lack the Adrb2 gene in the macrophages had blunted IL-6 and TF response, in comparison to Adrb2flox/flox mice after PM exposure [44]. Furthermore, PM exposure in mice with the depleted alveolar macrophages resulted in a decrease in both IL-6 release and thrombus formation [52]. These findings supported the results from in vitro studies which investigated PM induced inflammation through activation of the sympathetic nervous system via β2AR signaling in the macrophages. This further activated the release of IL-6, TNF-α, and TF leading to the thrombus formation [44][52][53][54][55][56][57][44,52,53,54,55,56,57].
PM may also induce oxidative stress, resulting in DNA damage [55][57][55,57]. Sirtuin 1 (Sirt1), the NF-ĸB regulatory gene, plays a key role in controlling the effects of PM-mediated inflammation [53]. Sirt1-knockout mice showed a higher level of inflammatory cytokines such as IL-6 and TNF-α after being exposed to PM2.5 [53], whereas a blunted inflammatory response was observed in the IL-6 knockout mice [52][56][52,56]. These findings emphasized the mechanistic links between the inhalation of PM and the stimulation of the sympathetic nervous system via β2AR, resulting in IL-6 release, systemic inflammation, and thrombosis, these processes being regulated by Sirt1. In addition, PM also induced oxidative stress in the rodent models. An increase in nitric oxide synthase (NOS) and heme oxygenase-1 (HO-1), and a decrease in catalase function in response to PM exposure were observed, together with an increase in the antioxidative response indicated by increase in glutathione (GSH) and a decrease in ascorbate level [55][57][55,57]. This increased anti-oxidative effect could be due to the compensation of the PM-mediated oxidative effects in those models.
PM2.5 may induce alveolar wall thickening and also enhance adhesion molecule and TF function [54]. PM2.5 also damaged vascular endothelial cells, resulting in TF release, which further activated the coagulation cascade and enhanced thrombus formation [53][54][55][56][53,54,55,56]. Intratracheal instillation of road tunnel dust in C57BL/6 mice could trigger TF release more extensively than in the mice exposed to urban dust, which was more pronounced at 48 h than at four hours after exposure [58]. These findings indicated that PM induced vascular injury and activated the TF pathway, with this process dependent on the composition of the PM and occurring in a time-dependent manner.
A study in hamsters revealed that PM2.5 exposure showed lower vWF levels, despite higher markers of vascular injury and vascular adhesion molecules [54]. This condition is characteristic of disseminated intravascular coagulation (DIC) which demonstrated obvious effects of PM2.5 on the prothrombotic state including evidence of extensive microvascular thrombi, decreased vWF, decreased coagulation factor levels, and prolonged clotting time, which resulted from a combination of the action of many clotting factors and vWF [54]. However, there are inconsistent reports. In a study in mice exposed to PM2.5 and PM10, no significant changes in vWF secretion and white blood cell (WBC) influx in the lungs and plasma were observed [59]. Although these two studies performed similar repetitive PM exposure for the same duration, these inconsistent findings could result from the differences in dose of PM exposure and in the species used. In the case of PM-mediated thrombosis, it has been shown that PM exposure could induce platelet activation, platelet function [59][60][59,60], and coagulation factors such as TF, and Factors II, VIII, and X [52][54][52,54]. Activation of platelets, TF, and coagulation cascades would further accelerate thrombin generation and induce a prothrombotic state [44][51][52][53][54][56][58][60][44,51,52,53,54,56,58,60]. One report revealed more thrombus formation at 48 h than at four hours after exposure, indicating that PM-accelerated thrombosis could be time-dependent [58].
The effect of PM on the fibrinolytic system remains controversial. Most studies reported that PM exposure alleviated fibrinolysis as a result of increased PAI-1 and decreased tPA mRNA expression, which caused the suppression of fibrinolytic activity and ultimately promoted a prothrombotic state [53][55][56][53,55,56]. A study which reported outcomes contradictory to these found that repetitive PM exposure increased tPA, reflecting the enhancement of fibrinolytic function [54]. It is possible that prolonged or chronic repetitive exposure to PM might induce extensive blood clots, leading to increased fibrinolytic activity as a compensatory response. However, further studies focusing on the fibrinolytic pathway are needed to enable us to understand the balance of each fibrinolytic factor and the ultimate effects of PM on fibrinolysis.
The effect of PM exposure on the blood cell count was inconclusive due to the conflicting data. Although PM was shown to increase the number of red blood cells (RBC) and hemoglobin (Hb) levels in mice [59], another study showed no significant impact [55]. The discordant results could be due to the difference in the duration of PM exposure, or the repetitive or prolonged duration of exposure, which might have more impact on PM-mediated changes than a single exposure or for a shorter duration. No WBC count changes were associated with PM exposure [55][59][55,59]. The effects of PM on platelet count were also uncertain since there were conflicting data among reports [52][54][55][59][52,54,55,59]. The possible explanation is that there were differences in particle type, exposure dose and/or duration, and in the severity of systemic activation of the coagulation pathway, especially the induction of DIC in those studies. Nevertheless, these in vivo studies emphasized that PM could induce lung injury via the stimulation of inflammation and oxidative stress, which further activates platelets and the coagulation system, leading to a hypercoagulable state. A summary of in vivo reports on the effects of PM on inflammation, oxidative stress, and the coagulation system is shown in Table 2.
Table 2. The effects of particulate matter on inflammation, oxidative stress, blood parameters, and hemostatic changes: Evidence from in vivo studies.
| Models | Exposure/Method | Results | Interpretation | References | ||
|---|---|---|---|---|---|---|
| Inflammation and Oxidative Stress | Coagulation and Adhesion Molecules | Blood Parameters | ||||
| Male Wistar rats 10–12 wk-old Cisplatin-induced AKI rats |
Intratracheal instillation of Cerium oxide nanoparticles (CeO2 NPs) 1 mg/kg |
Normal rats: # Kidney ↑ TNF-α, IL-6, GSH ↑ DNA damage # Lung tissue ↔ TNF-α, IL-6 ↓ catalase activity AKI rats: # Kidney ↑ TNF-α, IL-6, GSH ↑ DNA damage # Lung tissue ↑ TNF-α, IL-6 ↓ catalase activity |
Pulmonary exposure to CeO2 NPs induced inflammation and oxidative stress, and damaged DNA in the kidney. These effects were enhanced in kidney injury models. |
[57] | ||
| Male mice C57Bl6/j 8–12 wk-old IL-6+/+ IL-6−/− |
Inhalation exposure to concentrated ambient particles (CAPs) from downtown Chicago for 8 h/d for 3 d Evaluate at 24 h after exposure |
# IL-6+/+ vs. non-PM): Lung tissue ↑ IL-6/18s mRNA ↑ SP-B/18s mRNA BALF ↑ IL-6 ↑ TNF-α ↑ MCP-1 # IL-6−/−: Lung tissue ↓ IL-6/18s mRNA ↓ SP-B/18s mRNA BALF ↓ IL-6 ↔ TNF-α ↔ MCP-1 |
# IL-6+/+ vs. non- PM): Lung tissue ↑ TF/18s mRNA Plasma ↑ TAT complexes White adipose tissue ↑ PAI-1/18s mRNA # IL-6−/−: Lung tissue ↓ TF/18s mRNA Plasma ↓ TAT complexes White adipose tissue ↔ PAI-1/18s mRNA |
Exposure to all types of PM could activate inflammatory response, coagulation system and inhibit fibrinolysis, resulting in a prothrombotic state. PM-induced coagulation through IL-6 production and blocking IL-6 signaling could alleviate the thrombotic process. |
[56] | |
| Intratracheal instillation of urban PM (SRM1649a) 10, 100, 200 µg/animal Evaluate at 24 h after exposure |
# IL-6+/+ vs. non- PM): BALF ↑ protein ↑ macrophage, PMN ↑ IL-6 (dose-dependent) ↑ TNF-α # IL-6−/−: BALF ↔ protein ↔ macrophage, PMN ↓ IL-6 ↔ TNF-α |
# IL-6+/+ vs. non- PM): ↑ TF, ↑TF mRNA in lung tissue ↑ BALF D-dimer ↑ TAT complexes ↓ Bleeding time ↓ PT, ↓ PTT ↑ PAI-1/18s mRNA in the lung, adipose tissue ↑ PAI-1 in BALF # IL-6−/−: ↓ TF level, ↓TF mRNA in lung tissue ↓ BALF D-dimer ↓ TAT complexes ↔ PAI-1/18s mRNA in the lung, adipose tissue ↔ PAI-1 in BALF |
||||
| Male mice (C57BL/6) 8–12 wk-old |
Inhalation exposure to concentrated ambient particles (CAPs) from downtown Chicago for 8 h/d for 3 d |
↑ NE in the lung, BAT, adrenal gland ↑ IL-6 in BALF |
↑ TAT complexes ↑ thrombus formation ↓ thrombotic occlusion time |
Inhalation of PM caused catecholamine release and promoted IL-6-mediated thrombosis. | [44] | |
| Adrb1+/+Adrb2+/+ Adrb1−/−Adrb2+/+ Adrb1+/+Adrb2−/− Adrb1−/−Adrb2−/− |
Intratracheal instillation of urban PM (SRM1649a) 200 µg/animal Evaluate at 24 h after exposure |
BALF # Adrb1+/+Adrb2+/+ (vs. non-PM): ↑ IL-6 ↔ TNF-α, MCP-1 # Adrb1−/− Adrb2+/+ (vs. non-PM): ↑ IL-6 ↔ TNF-α, MCP-1 # Adrb1+/+Adrb2−/−: ↓ IL-6 ↔ TNF-α, MCP-1 # Adrb1−/−Adrb2−/−: ↓ IL-6 ↔ TNF-α, MCP-1 |
Plasma # Adrb1+/+Adrb2+/+ (vs. non-PM): ↑ TAT complexes ↓ thrombotic occlusion time # Adrb1−/− Adrb2+/+ (vs. non-PM): ↑ TAT complexes # Adrb1+/+Adrb2−/−: ↓ TAT complexes ↑ thrombotic occlusion time # Adrb1−/−Adrb2−/−: ↓ TAT complexes |
β2AR encoded by the Adrb2 gene in alveolar macrophages was necessary for PM-induced upregulation of IL-6, and enhanced susceptibility to thrombotic events. | ||
| Adrb1+/+Adrb2+/+ Adrb1+/+Adrb2−/− |
Inhalation exposure to concentrated ambient particles (CAPs) from downtown Chicago for 8 h/d for 3 d |
# Adrb1+/+Adrb2−/−: ↓ IL-6/18s mRNA |
# Adrb1+/+Adrb2−/−: ↓ TAT complexes ↓ TF |
|||
| Lyms-Cre Adrb2flox/flox mice (macrophage-specific deletion of β2AR) vs. Adrb2flox/flox |
Inhalation exposure to concentrated ambient particles (CAPs) from downtown Chicago for 8 h/d for 3 d Pretreated with formoterol (long-acting β2AR agonist) 1 × 10−5 M via inhalation twice every 12 h |
BALF # Adrb2flox/flox without formoterol: ↑ IL-6 in BALF # Adrb2flox/flox with formoterol: ↑↑ IL-6 # Lyms-Cre Adrb2flox/flox: ↓ IL-6 # Lyms-Cre Adrb2flox/flox with formoterol: ↓ IL-6 (vs. Adrb2flox/flox) ↔ IL-6 (vs. without formoterol) |
Plasma # Adrb2flox/flox without formoterol: ↑ TAT complexes ↑ factor II, TF mRNA ↓ thrombotic occlusion time # Adrb2flox/flox with formoterol: ↑ factor II, TF mRNA ↓ thrombotic occlusion time # Lyms-Cre Adrb2flox/flox: ↓ factor II, TF mRNA ↓ TAT complexes ↑ thrombotic occlusion time # Lyms-Cre Adrb2flox/flox with formoterol vs. Adrb2flox/flox: ↓ factor II, TF mRNA ↑ thrombotic occlusion time vs. without formoterol: ↔ factor II, TF mRNA ↔ thrombotic occlusion time |
|||
| Male mice C57Bl6/j Old mice (20 mo-old) vs. Young mice (10 wk-old) |
Inhalation of ambient PM2.5 and PM10 at the roadside tunnel for 25–26 d (A) tunnel-filtered (B) tunnel-exposed in urban roadside tunnel (C) control in clean facility |
# Young mice (vs. non-PM): ↔ WBC in BALF # Old mice (vs. young mice) in clean air: ↑ WBC in BALF # Old mice (vs. young mice) with PM: ↔ WBC in BALF |
# Young mice (vs. non-PM): ↔ lung vWF ↔ plasma vWF ↓ lung TM ↑ P-selectin ↔ PF4 # Old mice (vs. young mice) in clean air: ↑ lung VWF ↑ plasma VWF ↔ lung TM ↑ P-selectin ↔ PF4 # Old mice (vs. young mice) with PM: ↑ lung vWF ↔ plasma VWF ↔ lung TM ↔ P-selectin ↔ PF4 |
# Young mice (vs. non-PM): ↑ RBC, Hb ↑ platelets ↔ WBC # Old mice (vs. young mice) in clean air: ↑ RBC, Hb ↑ platelets ↑ WBC # Old mice (vs. young mice) with PM: ↔ RBC, Hb ↔ platelets ↔ WBC |
Continuous inhalation of particulate matter air pollution triggered inflammatory response, and activated platelets, and endothelial cells. The older mice had higher inflammatory biomarkers at baseline, therefore the PM-mediated effects were not demonstrated in the old mice. |
[59] |
| Male mice C57Bl6/j with spontaneous hypertension 11–12 wk-old |
Intratracheal instillation particulate matter # Road tunnel dust (RTD): 0.3, 1, 3, and 10 mg/kg # Urban dust (EHC-93) from Environmental Health Center in Ottawa, Canada 10 mg/kg Evaluation of lung tissue at 4, and 48 h after PM exposure |
# RTD (at 10 mg/kg): - at 4 h: ↑ TF ↑ thrombus formation - at 48 h: ↑ TF ↑↑ thrombus formation # EHC-93: - at 4 h: ↔ TF ↑ thrombus formation - at 48 h: ↑↑ TF ↑↑ thrombus formation |
PM induced procoagulant activity in the lungs, via increased TF expression and aggravated thrombus formation. | [58] | ||
| Hamsters (Pfd Gold) 100–110 g |
Intratracheal instillation of polystyrene particles: # 60 nm UFP -unmodified 500 μg/animal -carboxylated 500 μg/animal -amined 5, 50, 500 μg/animal # 400 nm: Amine-modified polystyrene particles 500 μg/animal Evaluation of BALF at 1 h after UFP exposure |
# Unmodified and carboxylated UFP: ↔ PMN influx # Amine-UFP (60 nm): ↑ PMN influx (50 and 500 µg/animal) ↑ protein, histamine (500 μg/animal) # Amine-particles (400 nm): ↑ PMN influx ↑ BALF protein ↔ BALF histamine |
# Unmodified and carboxylated UFP: ↔ thrombus formation # Amine-UFP (60 nm): ↑ thrombus formation (at 50 and 500 µg/animal) # Amine-particles (400 nm): ↔ thrombus formation |
Exposure to both positively charged UFP (60 & 400 nm) resulted in inflammation in the respiratory tract, but only the UFP (60 nm) rapidly activated the clotting system within an hour, leading to thrombosis. | [51] | |
| Hamster 100–110 g |
Intratracheal instillation of polystyrene particles: # 60 nm UFP - unmodified 500 μg/animal - carboxylated 500 μg/animal - amined 5, 50, 500 μg/animal # 400 nm amined- polystyrene particles 500 μg/animal Evaluation of BALF at 1 h after UFP exposure |
# Unmodified and carboxylated UFP: ↔ PMN influx # Amine-particles (60 nm and 400 nm): ↑ PMN influx (50 μg) ↑↑ PMN influx (500 μg) |
# Unmodified and carboxylated UFP: ↔ thrombus formation # Amine-particles (60 nm): ↑↑ thrombus formation (50 μg) ↑ thrombus formation (500 μg) # Amine-particles (400 nm): ↔ thrombus formation |
UFP induced pulmonary inflammation and promoted thrombosis, but the degree of lung inflammation did not show a correlation with the extent of thrombosis. | [60] | |
| Intratracheal instillation of DEP (SRM 1650) 5, 50, 500 μg/animal Evaluate at 1 h after UFP exposure |
BALF ↑ PMN influx ↑ protein ↑ histamine (at 50 and 500 μg/animal) |
↑ thrombus formation (50 μg) ↑↑ thrombus formation (500 μg) ↓ PFA100 closure time |
DEP exposure activated platelet and thrombin generation, leading to thrombosis. | |||
| Female mice (C57BL/6) 8–10 wk-old sex-age-matched Sirt1 +/+ Sirt1 −/− Sirt1 overexpression in WT mice (vs. WT mice) |
Intranasal instillation of PM2.5 (SRM 8785) 100 µg/animal for 24 h |
# Sirt1 +/+: ↑ lung NF-ĸB ↑ BALF albumin, PMN ↑ BALF TNF-α & IL-6 # Sirt1 −/−: ↑↑ lung NF-κB ↑↑ BALF albumin, PMN ↑↑ BALF TNF-α & IL-6 |
# Sirt1 +/+: ↑ lung fibrin formation ↓ TFPI ↑ TF ↑ lung PAI-1 ↔ plasma PAI-1 ↓ lung TM # Sirt1 −/−: ↑ ↑ lung fibrin formation ↓ ↓ TFPI ↑ TF ↑ ↑ lung PAI-1 ↔ plasma PAI-1 ↓↓ lung TM # Sirt1 overexpression: ↓ lung fibrin formation ↑ lung TM |
PM2.5 exposure promoted pulmonary vascular injury and enhanced inflammation, coagulation, and inhibited fibrinolysis, which was regulated by Sirt1 and NF-κB pathways. | [53] | |
| Male SD rats 8–12 wk-old |
Intratracheal instillation of PM2.5 once every 3 d for 30 d Doses: - Low dose: 1.8 mg/kg - Middle dose: 5.4 mg/kg - High dose: 16.2 mg/kg PM2.5 was collected from central Beijing, China |
↑ Alveolar wall thickening ↑ IL-6, IL-1β, CRP ↔ MCP-1 |
↓ Aortic valve peak blood flow ↑ thrombus formation ↑ TF ↑ TAT complexes ↑ Factor Xa ↑↑ D-dimer ↓ TM ↔ TFPI ↑ tPA ↓ vWF ↑ PT, PTT, TT ↔ fibrinogen ↑↑ ICAM-1, VCAM-1 |
↓ platelets | PM2.5 induced vascular endothelial injury, systemic inflammatory response, altered coagulation factors, anticoagulant pathway, and fibrinolytic system, resulting in the prothrombotic state, and DIC. | [54] |
| Male Wistar Kyoto (WKY) rats 12–15 wk-old |
Intratracheal instillation of PM2.5 and PM10 from The Northern and Southern Mexico - Total fraction - Insoluble fraction - Soluble fraction (control) of each PM2.5 and PM10 3.3 mg/kg Evaluation at 24 or 72 h after PM exposure |
# Total fraction and insoluble fraction of PM2.5 & PM10: ↑ BALF cell count ↓ alveolar macrophages Lung tissue ↑ total protein, ↑ albumin, ↓ ascorbic acid ↑ MIP-2, TNF-α mRNA ↑ BALF MIP-2, TNF-α ↑ HO-1 ↑ LOX-1R, ↑ NOS |
# Total fraction and insoluble fraction of PM2.5 & PM10: ↑ lung TF mRNA ↓ tPA mRNA ↑ PAI-1 mRNA |
# Total fraction and insoluble fraction of PM2.5 & PM10: ↔ RBC, Hb, Hct, platelet, and WBC |
Exposure to PM aggravated pulmonary inflammation and oxidative stress, as well as disruption in the procoagulant and fibrinolytic pathways of the lung. | [55] |
| Male mice (C57BL/6) 8–12 wk-old IL-6+/+ IL-6−/− IL-6+/+ depleted alveolar macrophages |
Intratracheal instillation of PM10 from ambient air in Düsseldorf, Germany 10 μg/animal for 24 h # Pretreated with Intratracheally instillation of liposomal clodronate 120 mg/animal for 48 h before PM exposure (Setting of WT mice depleted of alveolar macrophages) |
BALF # IL-6+/+ vs. non-PM10: ↑ macrophage, PMN ↑ IL-6, TNF-α, IFN-γ ↔ MCP-1, IL-10, IL-12 # IL-6−/− vs. non-PM10: ↑ macrophage, PMN ↔ IL-6 ↑ TNF-α ↔ MCP-1, IL-10, IL-12, IFN-γ # IL-6−/− vs. IL-6+/+: ↓ IL-6 ↔ TNF-α, MCP-1, IL-10, IL-12, IFN-γ # IL-6+/+ depleted alveolar macrophages: ↓ macrophage ↔ PMN ↓ IL-6 ↔ TNF-α, MCP-1, IL-10, IL-12, IFN-γ |
Plasma # IL-6+/+ vs. non-PM10: ↑ Factor II, VIII, X ↑ Fibrinogen ↓ Bleeding time ↓ PT, ↓ PTT ↓ thrombotic occlusion time ↑ TAT complexes # IL-6−/− vs. non-PM10: ↔ Factor VIII ↔ Bleeding time ↔ PT, ↔ PTT ↔ thrombotic occlusion time ↔TAT complexes # IL-6+/+ depleted alveolar macrophages: ↓ Factor VIII ↑ Bleeding time ↑ PT, ↑ PTT ↓ TAT complexes ↑ thrombotic occlusion time |
# IL-6+/+ vs. non-PM10: ↑ Platelet # IL-6−/− vs. non-PM10: ↔ Platelet # IL-6+/+ depleted alveolar macrophages: ↓ Platelet |
PM10 exposure-induced pulmonary inflammation, and IL-6 release. IL-6 was the key mediator, which enhanced coagulation factor function, resulted in shortening of clotting time, and led to thrombosis. Blocking either the macrophage function or IL-6 signal could alleviate PM-induced prothrombotic state. |
[52] |
AKI: acute kidney injury, BALF: bronchoalveolar lavage fluid, BAT: brown adipose tissue, β2AR: adrenergic receptor beta-2, CAPs: concentrated ambient particles, CeO2 NPs: Cerium oxide nanoparticles, CRP: C-reactive protein, d: days, DEP: diesel exhaust particles, DIC: disseminated intravascular coagulopathy, DNA: deoxyribonucleic acid, EHC-93: Environmental health center-93, GSH: glutathione, h: hours, Hb: hemoglobin, Hct: hematocrit, HO-1: heme oxygenase-1, ICAM-1: intercellular adhesion molecule-1, IL-1β: interleukin-1beta, IL-6: interleukin-6, IL-10: interleukin-10, IL-12: interleukin-12, IFN-g: interferon-g, LOX-1R: lectin-like oxidized low-density lipoprotein receptor-1, MCP-1: monocyte chemoattractant protein-1, MIP-2: macrophage inflammatory protein-2, mo: months, mRNA: messenger ribonucleic acid, NE: norepinephrine, NF-κB: nuclear factor-κB, NOS: nitric oxide synthase, PAI-1: plasminogen activator inhibitor-1, PFA100: platelet function analyzer-100, PF4: platelet factor 4, PM: particulate matter, PM2.5: particulate matter in diameter <2.5 µm, PM10: particulate matter in diameter <10 µm, PMN: polymorphonuclear cells, PT: prothrombin time, PTT: activated partial thromboplastin time, RBC: red blood cells, RTD: road tunnel dust, SD rats: Sprague-Dawley rats, SP-B: surfactant protein B, SRM: standard reference material, TAT complexes: thrombin-antithrombin complexes, TF: tissue factor, TFPI: tissue factor pathway inhibitor, TM: thrombomodulin, TNF-α: tumor necrotic factor-α, tPA: tissue plasminogen activator, TT: thrombin time, UFP: ultrafine particle, VCAM-1: vascular cell adhesion molecule-1, VWF: von Willebrand factor, WBC: white blood cells, wk: week, WT mice: wild type mice.