Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by maria cristina montresor and Version 2 by Rita Xu.
The tumor microenvironment (TME) is comprised of different cellular components, such as immune and stromal cells, which co-operate in unison to promote tumor progression and metastasis. In the last decade, there has been an increasing focus on one specific component of the TME, the stromal component, often referred to as Cancer-Associated Fibroblasts (CAF). CAF modulate the immune response and alter the composition of the extracellular matrix with a decisive impact on the response to immunotherapies and conventional chemotherapy. The most recent publications based on single-cell analysis have underlined CAF heterogeneity and the unique plasticity that strongly impact the TME.
- cancer-associated fibroblasts (CAF)
- stroma
- tumor microenvironment (TME)
1. Introduction
The Tumor Microenvironment: A Focus on Stromal Cells
Tumor cells are only one of the many partners involved in tumor development and progression. Indeed, the importance of the tumor microenvironment (TME), referred to as the cells and interactions that exist around the tumor cells that play a key role in supporting tumor growth, is now widely acknowledged. This concept initially started in 1889 with the seed and soil theory, where the English surgeon Stephen Paget, in “The distribution of secondary growths in cancer of the breast”, proposed the existence of a “congenial soil” that nourishes and fosters growth and survival of the metastatic tumor [1]. Yet, it was only in the early 1970s that pioneer researchers with an integrated view of the tumor, including immunity and angiogenesis, published their work. The first immunological studies reported the capacity of T lymphocytes, natural killer (NK) cells and macrophages to infiltrate the tumor mass [2][3][4][5][6][7][2,3,4,5,6,7]; as well, the presence of immunoglobulin and complement proteins was observed in the TME [8][9][8,9]. Judah Folkman, among others, discovered the process of angiogenesis in cancer [10][11][10,11] and a dependency between tumor growth and neo-angiogenesis was suggested [11][12][11,12].
In the last two decades, however, the focus has been restructured to include other TME components. The cells present in the TME can be roughly categorized into two types, those that are present in the parenchyma of healthy tissue before the tumor develops, and those that are recruited by the tumor itself. Indeed, the source of stromal cells in the TME is still debated and a large spectrum of precursors has been proposed [13][14][13,14].
In some tumors, such as pancreatic cancer, there is evidence of stromal cells that derive from either bone marrow–mesenchymal stromal cell (BM–MSC) [15][16][17][15,16,17], or from endothelial cells [18]. Studies performed in other cancer models suggest that the stromal component present in the TME may also result from the differentiation of resident fibroblasts [19], adipocytes [20], adipose-derived MSC (AD-MSC) [21][22][21,22], hematopoietic stem cells (HSC) [23][24][23,24], pericytes [25][26][25,26], and epithelial cells undergoing epithelial-to-mesenchymal transition (EMT) [27][28][27,28], as shown in Figure 1A. Overall, all of these cell types become part of the TME and are influenced by the tumor cells in a dualism where both influence each other, contributing to build what is the TME. Indeed, stromal cells can either exert an antitumor effect through the release of specific factors, such as transforming growth factor beta (TGF-β) [29], or they can prime the TME by promoting tumor growth through immune evasion, stimulating neo-angiogenesis, as well as also supporting the process of tumor proliferation and metastasis. This tumor-supporting stromal component is nowadays mostly referred to as cancer-associated fibroblasts (CAF) [13][14][13,14].
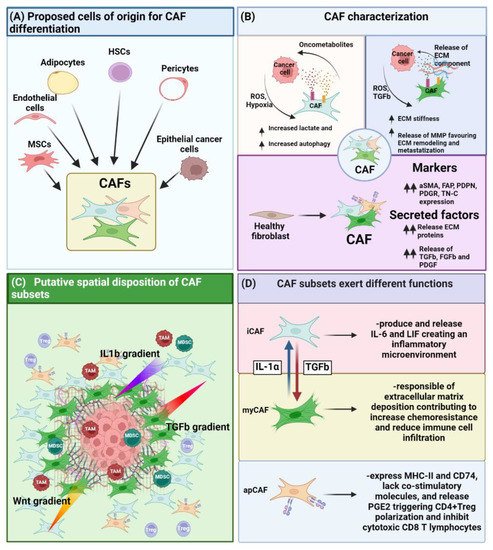
Figure 1. Role of CAF on the TME. (A) CAF can differentiate from various cell types such as MSC, endothelial, adipocyte, HSC, pericyte and epithelial cancer cells upon EMT. (B) CAF have been characterized under different point of views, ranging from their metabolic impact on the TME, their ability on altering ECM stiffness, as well as by the phenotypical and functional point of view enlisting a series of markers as well as factors they must secrete. (C) Different CAF subsets present a differential spatial disposition inside the TME. This effect is strictly dependent on the release by the tumor as well as by the immune cells of cytokines or growth factor, creating a gradient of differentiation triggering either the commitment towards myofibroblast-like CAF (myCAF) (TGF-β gradient), inflammatory CAF (iCAF) (IL-1β/Wnt gradient,). (D) Different CAF subsets exert different function.
2. The Emerging Heterogeneity within the TME Allows to Distinguish Functionally Different CAF
The term CAF generally refers to stromal cells present in the TME of solid tumors characterized by a distinct phenotype and functional and spatial patterns.
Within the tumor tissue, fibroblast activation relies on factors released by tumor cells and by tumor-infiltrating immune cells. These factors include, but are not limited to, TGF-β, platelet-derived growth factor (PDGF), and fibroblast growth factor 2 (FGF2) [13][30][13,30]. Altogether, the factors released in the TME may contribute to the acquisition of a pro-inflammatory gene signature. Indeed, these factors are responsible for driving the differentiation of stromal cells either residing in the tumor area or recruited to acquire functional and phenotypical features attributable to the CAF component, thus inducing a tumor-promoting phenotype in these cells [31].
Several researchers have attempted to identify shared criteria in order to build the basis for studying CAF starting from some common points.
The recent consensus paper published by Sahai and colleagues describes CAF as: (i) negative for epithelial, endothelial and lymphocytic markers such as E-cadherin, CD31 and CD45, respectively, and (ii) positive for markers attributable to a state of fibroblast activation, such as α-smooth muscle actin (αSMA) and fibroblast activation protein (FAP) [14]. Indeed, CAF have been identified both in vitro and in vivo based on proteins related to their activation status [13]. Over the years, these features have been complemented by the analysis of the expression of particular proteins involved in extracellular matrix (ECM) synthesis or modification, such as collagen type I and II, tenascin C, metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs) [13]. Furthermore, CAF modify the TME through the release of factors such as TGF-β, as well as PDGF and FGFs directly affecting the differentiation of immune and stromal progenitor cells recruited in the tumor area (Figure 1B).
However, one major obstacle in the identification of a pan-marker for activated fibroblasts is represented by the marked heterogeneity of fibroblast phenotypes across different types of tumors. Indeed, to date, there are no biomarkers exclusively expressed by CAF, but rather molecules that are indicative of a change of functional state [14]. The consensus paper by Sahai and colleagues defines CAF by the combined expression of TGF-β and αSMA [14][32][14,32].
In the last decade, innovative approaches such as single-cell technology have made it possible to bring greater clarity on the complexity of CAF. Indeed, the transcriptional expression profile of CAF at a single-cell resolution has highlighted the great heterogeneity present within CAF, and to also acknowledge the uniqueness of each tumor type in this context [33][34][35][33,34,35]. A comparison between breast and pancreatic cancer [36] highlighted the presence of activated fibroblasts with a distinct phenotypic profile, and partially overlapping with that of normal fibroblasts [37]. Evidence of stromal cell variability in cancer emerged from a comparison of the transcriptomic profile of CAF from breast cancer with that of healthy controls via single-cell RNA sequencing (scRNAseq), whereby the presence of four distinct populations of fibroblasts inside were identified in the lymph node of the patients characterized by different metastatization ability [38]. Another study reported the presence of two distinct populations of CAF in 3D cultures of pancreatic ductal adenocarcinoma, one with myofibroblast-like morphology named myCAF, and the other one characterized by the expression of inflammatory cytokines, named iCAF [39]. Specifically, the authors reported the presence of two subsets of CAF characterized by either a myofibroblastic or an inflammatory profile, whose functional characterization correlated with the patient’s clinical response. In both subsets, the differences observed were attributable to differential modulation of TGF-β signaling [39]. Interestingly, it has been reported the possible transition between myCAF and iCAF phenotypes in a cell contact fashion [39], highlighting the plasticity of CAF. Indeed, while the differentiation of myCAF is contact-dependent, that of iCAF may occur in absence of contact with cancer cells. Similarly, functional heterogeneity was also reported in non-small-cell lung cancer, where three CAF subpopulations that differentially expressed hepatocyte growth factor (HGF) and fibroblast growth factor 7 (FGF7), were reported. The first subset characterized by high HGF expression and variable FGF7 exerted a pro-tumor effect suppressing the immune response. The second subset highly expressed FGF7 and was moderately protective of cancers, by moderately suppressing the immune system, while the last subset, presenting low level of FGF7 and HGF, scarcely suppressed the immune system [40].
At the moment, three distinguishable fibroblast subpopulations inside the TME are reported which can be principally classified as: myCAF, iCAF and antigen presenting CAF (apCAF) (Figure 1D).
2.1. MyCAF and Their Role in ECM Remodelling
myCAF represent the major CAF subset present in many tumors, and these cells have been found close to the tumor mass where they perform opposite functions [41][42][41,42]. Cytoskeleton proteins such as αSMA and vimentin (Vim) are considered specific to determine myCAF in combination with TGF-β expression and ECM-related proteins. On one side, factors released by myCAF generate a stiff TME, thus favoring, for example, tumor metastasis and chemoresistance [43][44][45][43,44,45]. On the other hand, TGF-β can activate multiple pathways leading to CAF activation [46][47][46,47]; for example, it can trigger the activation of sonic hedgehog (SHH) signaling in CAF, thus increasing ECM deposition [48]. Indeed, activated fibroblasts release various types of collagens, laminins, fibronectins, proteoglycans, periostins, and tenascin C that favor tumor progression and metastasis [49]. Moreover, myCAF have been shown to produce proteases, such as different members of the MMPs [47][48][47,48] that digest the ECM, thus facilitating invasion of tumor cells into the nearby tissue. For example, stromelysin 1 is produced by activated fibroblasts and acts by cleaving E-cadherin and consequently promoting a premalignant phenotype in mammary epithelial cells, characterized by EMT and invasiveness [50][51][50,51]. Furthermore, myCAF are able to guide the direct invasion of cancer cells by means of a CAF-cancer cell co-migration mechanism [52], thus suggesting also a role for CAF in governing metastasis.
2.2. iCAF Are Responsible for the Creation and Maintenance of an Inflammatory TME
iCAF are inflammatory fibroblasts distinguished from other subtypes by their expression of the C-X-C motif chemokine 12 (CXCL-12) and interleukin-6 (IL-6) and of the inflammatory macrophage identification marker lymphocyte antigen 6 (Ly6C) [39][53][39,53]. Conversely to myCAF that are located close to the tumor mass, iCAF are spatially located at the periphery of the tumor, and their differentiation is induced by interleukin-1 (IL-1) released from nearby macrophage populations (Figure 1C) [39][53][39,53]. iCAF, in turn, can release leukemia inhibitory factor (LIF), which is responsible for maintaining an inflammatory state [53] and inducing the recruitment of tumor-associated macrophages (TAMs) [54].
TGF-β1 stimulation was shown also to indirectly induce iCAF in esophageal squamous cell carcinoma (ESCC). Indeed, TGF-β1 stimulates tumor cells to release the chemokine (C-X-C motif) ligand 1 (CXCL-1), which, in turn, has been observed to correlate to iCAF formation [55]. Conversely, others reported the capacity of TGF-β to inhibit Interleukin 1 receptor-like 1 (IL1-R1) expression, thus antagonizing interleukin 1 alpha (IL1-α) responses and blocking iCAF generation [56].
A bioinformatic deconvoluted analysis was shown to discern CAF subtypes providing new information on CAF phenotype. Indeed, in this study, the authors identified several genes associated with myCAF and iCAF in bladder carcinoma (BLCA), squamous cell cancer in the head and neck region (HNSC), and lung adenocarcinoma (LUAD), including 4 overlapping genes between myCAF and iCAF. Notably, gene ontology analysis reported that genes co-expressed in both myCAF and iCAF were involved in the formation of ECM [57]. Furthermore, in this study, an elevated presence of myCAF correlated to poor prognosis in different types of cancer [57]. The poor prognostic value of myCAF has been reported also by others [58][59][58,59], and related to the ability of myCAF to decrease the sensitivity to specific antineoplastic drugs, therefore worsening disease prognosis. On the contrary, the role of iCAF is still unclear, as their presence does not determine a significant difference in drug sensitivity, but rather has been associated with a better prognosis regardless of their number [57].
2.3. An Obscure Subset of CAF: Antigen Presenting CAF (apCAF)
In addition to myCAF and iCAF, another population of CAF has been identified and termed antigen-presenting CAF (apCAF) [60]. These cells express major histocompatibility complex II (MHC-II) and CD74, lack the canonical co-stimulatory molecules, and release prostaglandin E2 (PGE2). On one hand, the concomitant lack of costimulatory molecules and release of PGE2 was shown to enhance the ability of apCAF to promote the expansion of CD4+ regulatory T cells (Tregs) [60]. On the other hand, apCAF were shown to activate CD4 and CD8 T lymphocytes present in the tumor tissue in an antigen-specific manner, thus enriching their phenotype with diverse immunomodulatory functions [60][61][60,61]. The expression of programmed death-ligand 2 (PDL2) and Fas ligand (FasL) on these cells allows them to inhibit the cytotoxic activity of CD8 lymphocytes. Indeed, the use of monoclonal antibodies to neutralize the in vivo interaction between CD8 and PDL2 or FasL expressed on CAF was shown to restore the cytotoxic action of T lymphocytes [62].