Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Conner Chen and Version 1 by Philippe Gasque.
Mesenchymal stem cells (MSCs) play a critical role in response to stress such as infection. They initiate the removal of cell debris, exert major immunoregulatory activities, control pathogens, and lead to a remodeling/scarring phase. Interestingly, many viruses and particularly those associated to chronic infection and inflammation may hijack and polarize MSC’s immune regulatory activities to their own advantages. Virus will remain in the MSC perivascular niche while being protected from immune attack. In the context of immunodepression (e.g. organ transplantation) the hidden viruses may rebound and causing tissue injuries.
- innate immunity
- immune-regulation
- neural crest
- COVID-19
- chikungunya
1. MSC Viral Infection and Host Responses
Mesenchymal stem cells (MSCs) as aforementioned have mostly critical roles during injury, participating in immunomodulation and tissue repair. In addition, their perivascular location in multiple organs makes them potential targets for viral infection, as summarized in Table 1.
Table 1. Examples of viruses targeting MSCs and related pathologies.
| Cells | Viruses | Related Outcomes |
|---|---|---|
| Bone marrow-MSC | HIV | Inability to support Hematopoietic Stem Cells expansion and implication in HIV-related cytopenia [27][1]; HIV-related reactivation [187,188,189][2][3][4] |
| HCMV | Transmit to neighboring cells after reactivation [190,191][5][6] | |
| Modifies the physiological interaction between bone marrow (BM-MSCs and )-derived MSCs (BM-MSC) and hematopoietic stem cell (HSC) | ||
| Impairment of osteoblast regeneration, cartilage regeneration, hematopoiesis and properties of immune progenitor cells [191][6] | ||
| HHV | Lower proliferation rates and altered phenotypes related to malignant transformation [190,192,193][5][7][8] | |
| Influenza A H5N1 | Risk of transmission during bone marrow transplantation [194,195][9][10] | |
| RSV | Alteration of immunoregulatory functions [196][11] | |
| ZIKV | Impaired osteoblast differentiation and possible implication in development of bone pathologies [197][12] | |
| HBV | HBV-associated myocarditis and other HBV-related extrahepatic diseases [198][13] | |
| Lung resident MSC/pericytes | SARS-CoV-2 | Pericyte apoptosis and loss in COVID-19 patients [199,200][14][15] |
| SIV/HIV | Development of HIV-related pulmonary complications [197,201][12][16] | |
| Hepatic Stellate Cell (HepSC, Ito cells) | HIV | HepSC activation and chemotaxis through HIV gp120 envelope protein [202,[17203]][18] |
| HCV | HCV proteins as well as RNA released by hepatocytes are activating HepSC [204,205][19][20]. Constant activation leading to liver fibrosis [206,207,208,209][21][22][23][24] | |
| HBV | Release of IL-17 by infected cells which stimulate liver fibrosis by activation of HepSC [210,[25211]][26]. | |
| Mesangial Cell (Kidney) | HIV | HIV-associated glomerulosclerosis due to increased proliferation and matrix synthesis [212,[27213]][28] |
| HCMV | Glomerulosclerosis [214,215,216,217][29][30][31][32] | |
| HCV | Glomerulonephritis [218][33] | |
| ZIKV | Viral reservoir (persistent viruria) [219][34] | |
| SARS-CoV-2 | Stromal (MSC-like cells) are infected and may contribute to kidney fibrosis in a model of spheroid cultures and to be correlated with kidney fibrosis in COVID19 patients [220][35]. | |
| Brain Pericytes (BP) | HIV | Decreased BPs coverage of blood brain barrier (BBB) associated with higher permeability [221,222][36][37] |
| IL-6 and PDGF-BB secretions concur in HIV-induced CNS damage and BBB disruption [221,223][36][38] | ||
| HCMV | Contribute to HCMV dissemination [224][39] | |
| CXCL8, CXCL11, CCL5, TNF-α, IL-1β and IL-6 secretion causing neuroinflammation [225][40] | ||
| JEV | IL-6 secretion leads to ZO-1 degradation and BBB impairment [226][41] | |
| Prostaglandin E2 (PGE2) and RANTES secretion by BPs recruit leukocytes to the site of infection. Associated with BBB impairment, this provoke leukocyte infiltration and major neuroinflammation [227][42] | ||
| Herpes Simplex Virus (HSV) | BBB impairment associated with leukocytes recruitment leading to major neuroinflammation [228][43] | |
| ZIKV | Brain abnormalities and BBB defect [229,[44230]][45] | |
| Osteoblasts | HIV (gp120) | TNF-α and impaired Wnt/β-Catenin signaling promote bone demineralization and reduced bone mass leading to osteopenia and osteoprosis [231][46] |
| RRV | Imbalance in RANKL/OPG ratio in favor of osteoclastogenic activities and bone loss [232,233][47][48] | |
| Chikungunya Virus (CHIKV) | Proinflammatory (IL-6) and pro-osteoclastic (RANKL) effects in infected cells [234][49] | |
| HCV | Associated with bone density hardening and osteosclerosis [235][50] | |
| Increased risk of osteoporosis [236][51] | ||
| Increased risk of fracture [237][52] | ||
| Impairment of RANKL/OPG ratio [235][50] | ||
| ZIKV | Impaired osteoblasts function triggering an imbalance in bone homeostasis and inducing bone-related disorders [197][12] | |
| MeV | Higher expression of several osteogenic markers and osteogenic differentiation [238][53] | |
| Otosclerosis [239][54] | ||
| Paget’s disease [240][55] | ||
| Schwann Cell (SC) | HIV | Dorsal root ganglion neurotoxicity, including axon and myelin injury [241][56] |
| HSV/VZV | The principal mechanism evoked for HSV-induced Guillain-BSarré Syndrome (GBS) is a molecular mimicry of viral proteins [242][57] | |
| HCMV | Probable molecular mimicry generating autoantibodies against moesin expressed by SCs [243,244,245,246,247][58][59][60][61][62] | |
| ZIKV | Possible direct viral pathogenic effect or a cell-mediated inflammation in pathogenesis of ZIKV-associated GBS [248][63] | |
WThe following contents will address the different viral infections that affect MSCs and the resulting innate immune response given that MSC are immunocompetent gatekeepers known to express pattern recognition receptors (PRRs, e.g., RLR, TLR) and downstream signaling molecules (e.g., NF-κB, IRF3/7) [20,99][64][65]. In parallel to macrophage polarization, two distinct subsets of MSC may exist in tissues (MSC1, proinflammatory and MSC2 anti-inflammatory) [249,250][66][67]. In response to acute tissue injury and the release of alarmins derived from cell debris (apoptotic cells), MSC have a type 1 phenotype and help to recruit lymphocytes to sites of inflammation using MIP-1a and MIP-1b, RANTES/CCL5, CXCL9, and CXCL10 and to promote the clearance of cell debris and tissue repair. If the injury is too important and associated to cell necrosis and high levels of Tumor NFecrosis Factor (TNF)-α and IFNnterferon (IFN)-β produced by monocytes and T cells, respectively, MSC adopt an immune-suppressive phenotype (MSC2) by secreting high levels of soluble immunoregulatory factors, including kynurenine (Indoleamine 2 3-dioxygenase (IDO) pathway), PGE2 (COX2 pathway), NO, TGF-β1beta 1 (TGF-β1), IL10, Hepatocyte Growth Factor (HGF), and hemoxygenase (HO), that suppress adverse T cell proliferation and possible autoimmune response. TGF and IL10 will further control adaptive immune responses by mobilizing FoxP3+ T regulatory cells. Interestingly, viruses (e.g., those stimulating the TLR3 pathway by Poly I:C)inosinic:polycytidylic acid (Poly I:C)) may promote MSC2 anti-inflammatory and immunosuppressive phenotype to their own advantages by limiting the adaptive immune responses [251][68].
2. MSCs a Gatekeper and/or Reservoir of Viruses
2.1. Bone Marrow-Derived MSC (BM-MSC)
BM-MSCs are among the first and best described subsets of MSCs [49][69]. Genetic line tracing experiments using either Nestin or GLI-1 promoters indicated that perivascular BM-MSC could be derived from neural crest (NC) embryonic tissues, expressed neuroglial markers, as well as the beta3 adrenergic receptors arguing for a plausible role of the sympathetic nervous system to control MSC functions [17,64,151][70][71][72]. Physiologically, BM-MSCs constitute a stromal cell niche for HSCs, supporting their stemness and education [17,64,252][70][71][73]. Due to their relative ease of access, they represent a powerful source of cells as much for the study of the properties of MSCs as for the investigation of new therapeutic avenues. Indeed, BM-MSCs are now widely used as treatment given their beneficial immunoregulatory properties. They are used as carriers for the delivery of miRs or protein factors with therapeutic activities, physiologically expressed by MSCs or induced following an adenoviral or lentiviral infection [253][74]. Additionally, BM-MSCs-derived exosomes have shown a similar therapeutic potential [134][75].
However, due to their proximity with the HSC and hematopoiesis processes, their impairment, in particular through a viral infection, could be critical for BM-related diseases. Moreover, given bone marrow engraftment is the unique suitable treatment for certain diseases (e.g., hematological malignancies), the persistence of virus-infected cells in grafts could represent a major risk, especially in patients that are usually immunocompromised.
BM-MSCs have been shown to be targeted by a variety of viruses during the natural history of a clinical infection.
Human Immunodeficiency Virus (HIV), that causes Acquired Immuno Deficiency Syndrome (AIDS), was shown to infect BM-MSC and integrate its DNA in BM-MSC’s genome [29,188][3][76]. Also, both intra- and extracellular interactions of HIV proteins Tat and Nef with BM-MSCs were observed [188,189][3][4]. This leads to an impaired osteoblastic/proadipogenic differentiation therefore promoting respectively decreased bone marrow density and fat toxicity described in HIV-infected patients [188,189][3][4]. Furthermore, HIV is able to induce senescence of BM-MSCs through its proteins Tat and Nef, but also p55-gag [27,189][1][4]. BM-MSC senescence has been associated to HIV infection-related cytopenia [27][1]. Finally, a role of BM-MSCs in HIV-related disease is the reactivation of HIV in latently-infected cells [187][2].
Herpesviridae is a large family that includes diverse viruses causing human diseases, such as Human Cytomegalovirus (HCMV), or Human Herpesviruses (HHV). Among these viruses, HCMV, HHV-1, HHV-3 (or Varicella-Zoster Virus, VZV) and HHV-8 (Kaposi’s Sarcoma-Associated Herpesvirus) exhibited the ability to infect BM-MSCs [30,190,192,193,254][5][7][8][77][78]. HCMV is a leading cause of congenital birth defects, as well as the major cause of diseases in immunocompromised individuals, notably following organ or BM transplant.
Of note, BM is an important site involved in the pathogenesis of chronic HCMV infection. The virus establishes latency in hematopoietic progenitors and can be transmitted after reactivation to neighboring cells. HCMV has deleterious effects on BM-MSCs function, changing the repertoire of cell surface markers expression (CD29, CD44, CD73, CD105, CD90, MHC class I and ICAM-1), and modifying the physiological interaction between BM-MSCs and HSC [191][6]. In addition, similarly to HIV, HCMV alters BM-MSCs biology and might contribute to the development of diseases (impairment of osteoblast regeneration, cartilage regeneration, hematopoiesis and properties/functions of immune progenitor cells), due to a deterioration of BM-MSC differentiation capabilities [191][6]. More strikingly, BM-MSCs were evoked as a potential reservoir for HCMV [30][77], which could be crucial for treatments involving BM engraftment.
HHV-8 also induced alterations of BM-MSCs, infected cells displaying lower proliferation rates and altered expression of Kaposi’s Sarcoma markers as well as altered phenotypes related to malignant transformations [192][7].
Even if BM-MSCs are physically less exposed to respiratory viruses, the influence of these viruses on BM-MSCs has been explored in several studies [194,195,196][9][10][11]. Avian Influenza A (H5N1) virus productively infects BM-MSCs provoking cell death and IL-6, CCL2 and CCL4 secretion by co-cultured monocytes [195][10]. The link between these findings and abnormal hematologic clinical descriptions such as lymphopenia, thrombocytopenia, and pancytopenia observed during avian flu remains to be addressed. Still, as for HCMV, the infection of BM-MSCs by Influenza virus might represent a risk of transmission during BM transplantation [194][9]. BM-MSCs contribute to Respiratory Syncytial virus-related lung disease. Indeed, this virus is able to infect and replicate in BM-MSCs, altering their immunoregulatory functions via an increase of IFN-β and IDO expression [196][11]. Zika virus (ZIKV) is a recently reemerging flavivirus, responsible for dengue-like syndrome in most of the cases but also associated with Guillain-Barré syndrome (GBS) in severe cases [230][45]. ZIKV, which is more known for its implication in neurological induced disorders (as described in Brain pericytes section), has the ability to infect and replicate in BM-MSCs [197][12]. BM-MSCs infection by ZIKV causes increased IL-6 expression and impaired osteoblast differentiation, pointed out by a decreased expression of alkaline phosphatase (ALP) and Runt-related transcription factor 2 (RUNX2) [197][12]. These data demonstrate the potential involvement of ZIKV in the development of bone pathologies. Finally, BM-MSCs might serve as an extrahepatic reservoir for Hepatitis B virus (HBV). Indeed, BM-MSCs are infected in vitro by HBV [198][13]. Moreover, BM-MSCs are able to transport HBV to injured tissues, as evidenced by transplantation of BM-MSCs in a mouse model of myocardial infarction, resulting in HBV infection of injured heart and other damaged tissues [198][13]. Thus BM-MSCs might play a critical role in HBV-associated myocarditis and other HBV-related extrahepatic diseases.
BM-MSCs are best known for their immunoregulatory activities during injury as aforementioned. They possess antiviral effector functions (Figure 31), even if the innate immune response of BM-MSCs against each virus previously cited has not been examined in great depth. BM-MSCs are basically expressing several cytosolic PRR albeit at low levels [10][79].
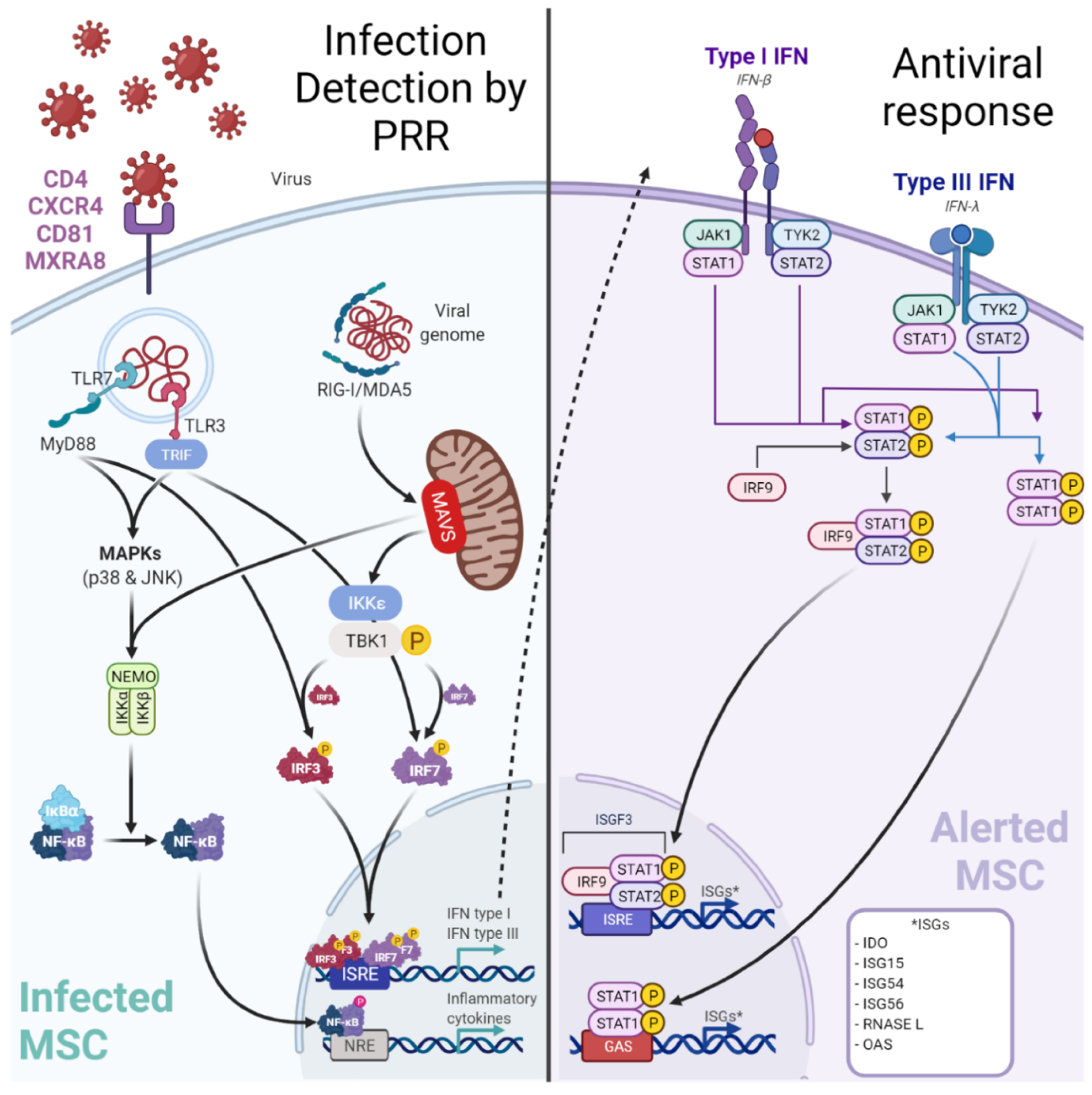
Figure 31. Infection of MSC by viruses may be controlled by a canonical innate immune response. Viruses may target perivascular MSC naturally expressing receptors (e.g., MXRA8/alphavirus) to grant entry. Among the up-regulated cytosolic pattern recognition receptors (PRRs), Retinoic acid-inducible gene-I (RIG-I)-like receptors (RLR: RIG-I, Melanoma-Differentiation-Associated Gene-5 or MDA5) and Toll-Like Receptor 3 (TLR3) are important to detect viral RNA. After sensing, MSCs engage different cell signaling pathways according to the stimulated PRR. A TLR3-dependent sensing activates mitogen-activated protein kinase pathways (through p38 MAPK and p46 JNK). RLR-dependent sensing stimulates IFN signaling pathway through TBK1/IKK-ε and subsequent interferon regulatory factor (IRF) 7 phosphorylation. These signaling pathways both trigger the production of pro-inflammatory cytokines and peptides with antiviral activities. Hence, in viral context MSCs express increased levels of IL-1β, IL-6, IL-8, IL-11, IL-12p35, IL-23p19, IL-27p28, TNF-α and CCL5/RANTES to recruit and activate adaptive immune cells (T/B lymphocytes). Furthermore, MSCs produce IFN-β and IFN-λ1). Classically, type I IFNs (such as IFN-β) induce the expression of Interferon Stimulated Genes (ISGs, e.g., RNASEL) by the interaction with the IFN receptor (IFNAR).
Yet in the context of a viral infection, it has been demonstrated (using Poly I:C to mimic RNA viruses) that BM-MSCs can up-regulate their PRR expression [255][80]. Among the up-regulated cytosolic PRR, Retinoic acid-inducible gene-I (RIG-I)-like receptors (RLR: RIG-I, Melanoma-Differentiation-Associated Gene-5 or MDA5), and Toll-Like Receptor 3 (TLR3) are important to detect viral RNA [255,256][80][81]. After sensing, BM-MSCs engage different cell signaling pathways according to the stimulated PRR. A TLR3-dependent sensing activate mitogen-activated protein kinase pathways (through p38 MAPK and p46 JNK) [256][81]. Whereas RLR-dependent sensing stimulates IFN signaling pathway through TBK1/IKK-ε and subsequent interferon regulatory factor (IRF) 7 phosphorylation [255][80]. These signaling pathways both trigger the production of pro-inflammatory cytokines and peptides with antiviral activities. Hence, in viral context BM-MSCs express increased levels of IL-1β, IL-6, IL-8, IL-11, IL-12p35, IL-23p19, IL-27p28, TNF-α, and CCL5/RANTES [255,256,257][80][81][82]. Furthermore, BM-MSCs produce IFN-β and IFN-λ1 in a RIG-I-dependent manner in contrast to other MSC subtypes producing it in both TLR3 and RIG-I dependent manner [255][80]. Classically, type I IFNs (such as IFN-β) induce the expression of Interferon Stimulated Genes (ISG) by the interaction with the IFN receptor (IFNAR). To date, the expression of ISGs has not been explored in BM-MSCs and needs further investigations. Interestingly, an antiviral activity of IDO has been demonstrated by decreasing HCMV and HSV-1 replication in BM-MSCs [258][83].
Of note, exosomes derived from allogenic BM-MSC (e.g., EXOFLOTM) have been used clinically to limit successfully the cytokine storm and the associated tissue injuries in patient with COVID-19 [259][84]. This would argue that MSC in tissues may be able to control the infection mediated by SARS-CoV-2.
2.2. Lung Resident-MSC and Viruses
To date, lung resident-MSCs (LR-MSCs) have been mostly studied in the context of fibrosis and bronchiolitis obliterans syndrome, notably after lung transplantation [52,260][85][86]. Several important and recent gene tracing studies argued for a critical role of perivascular MSC, derived notably from NC progenitors, in lung diseases [143,147][87][88]. However, LR-MSCs have been poorly studied in the virology field even if their pivotal position makes them potentially susceptible to viruses and particularly respiratory viruses. Supporting this, possible interactions between SARS-CoV-2 and MSC have been envisaged in COVID-19 positive patients experiencing pericytes loss and apoptosis (observed by cleaved-caspase 3 immunostaining) [199][14]. Because pericytes often include MSCs, as discussed above, these findings suggest LR-MSCs as potential targets of SARS-CoV-2 in the lung [200][15]. Currently available data regarding the expression of ACE2 and TMPRSS2 in MSCs are discordant [261][89].
LR-MSCs have shown the highest permissiveness, replication rate and release during HCMV infection in cultured cells [30][77]. Moreover, LR-MSC is now recognized as a natural reservoir for HCMV after its detection in seven out of nine individual donors [30][77].
Lung PDGFβ+ MSC were recently shown as targets for HIV. These cells express both the primary receptor of HIV (CD4) and its major co-receptors (CXCR4 and CCR5), permitting productive infection and replication of HIV in vitro [201][16]. This finding may indicate a possible implication of LR-MSCs in the HIV-related pulmonary complications [201][16].
Surprisingly, in comparison to advances made for other MSCs subtypes, very few data are available to date regarding the specific antiviral response mounted by LR-MSCs during infection. Efforts should be made to fill this gap since it has already been demonstrated that a different antiviral response may be mobilized by tissue-specific MSCs [255][80].
2.3. Adipose Stem Cells and Viruses
Adipose Stem Cells (ASCs) are a subtype of MSCs found in higher amounts than BM-MSCs and their isolation is easier, safer, less painful and less time consuming using liposuction techniques [50,262][90][91]. Consequently, they became an alternative to BM-MSCs for cell-based or exosome-based therapy using MSCs [263,264][92][93]. ASC can differentiate into different lineages and including into a Schwann-cell like phenotype expressing S100, glial fibrillary acidic protein (GFAP) with neurite outgrowth activity [265][94]. They are now studied to be used in a broad range of diseases [263][92]. MSCs are highly sensitive to their environment, influencing their responses through notably TLR or RLR [255][80]. ASCs have important immunomodulatory functions shown by their high cytokine response [255][80]. Notably, viral stimuli induce an important activation of these cells [255][80]. Furthermore, the importance of immunoregulatory function of ASC is highlighted by their capacity to produce various adipokines (adiponectin, leptin) that influence the inflammatory responses in most tissues [266][95]. Most of the studies regarding adipose tissue and viral infection have focused on adipocytes which are the main cell type of adipose tissue and also a derivative of ASCs. In the context of HIV, anti-viral treatments were shown to accumulate in adipocytes and alter their adipokine production, explaining some long-term complications of the therapies [267][96]. Adipocytes are mostly resistant to direct viral infection by Hepatitis CV Virus (HCV) or HBV [268,269][97][98]. Due to their differentiation capabilities and their accessibility, ASC represents a good candidate for in vitro cell infection model [268[97][98],269], research on factors influencing viral replication like miR-27a in HCV infection [269][98] or stem-cell based therapies during HBV chronic infection [268][97]. Nevertheless, further studies are needed to explore the direct role of ASC in the immune response in the course of viral infection and addressing the regulation in adipokine and miRNA production.
2.4. MSC of the Liver: Hepatic Stellate Cell/Ito Cell and Viruses
Hepatic Stellate Cells (HepSC, also named Ito cells) have been firstly described by Von Kupffer in the 19th century [70[99][100],270], for review, [13,18,271][101][102][103]. Using gene tracing experiments, they can be derived from myelin P zero (MPZ)+ NC cells and they express GFAP (astroglial marker) particularly in response to injuries (e.g., [12][104]). In physiological conditions, HepSCs are mostly long-lived quiescent cells residing in the perisinusoidal space of Disse [70,272][99][105]. They represent 15% of resident liver cells [273][106] and are fat-storing, vitamin-A rich, with presence of long processes [70,271,274][99][103][107]. These cells interact with neighboring cells like hepatocytes, resident macrophages (Kupffer cells), endothelial cells or nerves [70,273][99][106]. Also, they express markers of mesenchymal origin like desmin, alpha SMA [272,273][105][106]. Under pathological conditions, they show proliferative activity and differentiate in myofibroblast-like cells [70][99]. Activated HepSCs produce large amounts of extracellular matrix proteins (ECM) and matrix metalloproteases (MMPs) [70,273,275,276][99][106][108][109].
Quiescent HepSCs participate in physiological conditions to liver homeostasis, regeneration, development, retinoid metabolism, extracellular matrix homeostasis, and drug metabolism [277][110]. Upon liver injury, various stimuli, depending in the liver disease, activate HepSCs. The chronic activation of HepSC leads to excessive ECM accumulation and liver fibrosis [206,277][21][110].
In the context of viral infection, liver fibrosis represents a critical complication of end-stage liver disease progression. It mainly occurs through the persistent activation of HepSCs during chronic infection. This process has been studied during either HCV, HBV, or HIV pathologies.
HepSC activation occurs through direct or indirect viral effects [207][22]. HCV can directly activate HepSCs via either E2 protein binding to CD81 [208[23][110],277], NS3-NS5 proteins [209,277][24][110] or dsRNA [277,278][110][111]. HepSC expression of CCR5 and CXCR4 co-receptors for HIV has been described [202][17]. Moreover, HepSC infection by HIV has been established in vitro but not in vivo [202,203][17][18]. HIV through its envelope protein gp120 has been shown to induce HSC activation and chemotaxis [202][17].
The indirect HepSC activation also represents an important mechanism. In fact, the hepatic inflammatory environment is an important factor of HepSC activation thus leading to pro-fibrogenic factor release [277][110]. Hepatocytes produce profibrogenic factors like TGF-β1 or TIMP-1 [204[19][110],277], apoptotic bodies [277][110] or ubiquitin carboxy-terminal hydrolase L1 (UCHL1) [205][20] during HCV infection. Also, activated HepSC will release cytokine (i.e IL-1α) which will further stimulate hepatocytes to produce pro-inflammatory cytokines (IL-6, IL-8) [277][110]. In the course of HBV infection, lymphocytes are implicated in inflammatory tissue damages [210][25]. As shown for the hepatocytes, activated HepSCs may participate in the activation of immune cells thus amplifying the local inflammatory environment. For example during HBV infection, HepSCs could promote Th17 cell activation through IL-17RA-dependent proinflammatory cytokine expression [211][26]. Similarly, during HIV chronic infection, many factors participate in HepSC activation and fibrogenesis like natural killer (NK) cells dysfunction, intestinal microbial translocation, Kupffer cell inflammatory response, hepatocytes apoptosis, and liver damages [202][17].
The implication of HepSCs in the context of liver fibrosis during chronic viral diseases has been highly studied. However, their role during acute hepatotropic viral infection notably the importance of their immune response for viral clearance has been less explored.
2.5. MSC of the Kidney: Mesangial Cell and Viruses
Mesangial cells (MCs) is the name used to designate the resident perivascular MSCs of the kidney and chiefly involved in fibrosis [4,144,147][88][112][113]. They are derived from the NC and responsible for mesangium matrix formation and glomerular capillaries support [12][104]. As for other MSCs, their location at the periphery of the vessels allows them to act as gatekeepers and to sense their environment in case of injury such as viral infection [14][114]. Similar to what is observed in other organs, viral infection in the kidney may lead either to a direct deleterious viral effect on targeted cells or to an indirect effect, due to an inappropriate immune response. MC participates in both mechanisms. Indeed, MCs were demonstrated to be targeted by various viruses [212,214,219,279,280,281,282,283][27][29][34][115][116][117][118][119]. First, HIV was shown to infect MCs [212][27] with an orphan G protein-coupled receptor 1 (GPR1) as a co-receptor [213][28]. Additionally, HIV affects MCs either directly or indirectly leading to proliferation and matrix synthesis, thus MCs play a role in HIV-associated glomerulosclerosis [28][120]. HCMV is the most threatening pathogen after a kidney transplantation, due to immunosuppression [284][121] and potential virus rebound in immunocompromised host patient. HCMV is associated with the development of glomerulosclerosis due to the matrix deposition caused by MCs [214,215][29][30]. Of note, MCs were shown to be targeted by HCMV and to allow its efficient replication [216[31][32],217], even if the link between this infection and the pathology associated is poorly understood [214][29]. Among the viruses of critical interest, HCV is a major problem worldwide, despite recent therapeutic improvements [285][122]. The pathology associated with HCV is mostly liver disease, but with frequent extrahepatic complications, such as glomerulonephritis. HCV triggers TLR3 activation of MC leading to the release of procoagulant factors that causes vascular thrombosis and finally glomerulonephritis [279][115]. Furthermore, MCs may be infected by ZIKV. It has been demonstrated that these cells may serve as a reservoir for the virus, thereby this finding may explain the high level persistent viruria observed [219][34]. However, to date, no link has been established between ZIKV infection of MCs and kidney disease.
Of note, the antiviral response of MCs was not studied in the context of the viral infection aforementioned. However, viral RNA and DNA trigger a common antiviral response in MCs notably through RIG-I, MDA5 and TLR3 [286,287,288][123][124][125]. This response includes notably Interferon-type I response and secretion of proinflammatory cytokines and chemokines.
On the other hand, MCs also participate in inappropriate immune responses in case of infection. Indeed, immune complexes associated with viral RNA or DNA triggers overwhelmed immune response leading to glomerulonephritis. This mechanism was notably shown in a mouse model of Immunoglobulin A nephropathy (IgAN) induced by Sendai virus [280][116], but also in patients suffering from Lupus nephritis [289][126].
2.6. Brain MSC: Brain Pericytes and Viruses
Brain pericytes (BPs) are the resident MSCs of the brain [68][127]. BPs are essentially derived from NC [11,15][128][129]. With endothelial cells, and astrocytes, they constitute the blood-brain-barrier (BBB), the specialized vascular unit of the brain, responsible for its protection from external factors. Due to this major role in brain defense, an infection of BPs could lead to BBB disruption and increased permeability. BPs are well-equipped to ensure their sentinel function. They were shown to express PRR, notably TLR9, allowing them to be responsive in case of microbial infection, particularly by non-canonical inflammasome activation [290][130]. Moreover, BPs are immunocompetent cells with NO, IL-1β, IL-3, IL-9, IL-10, IL-12, IL-13, TNF-α, IFN-γ, G-CSF, GM-CSF, Eotaxin, CCL3, and CCL4 secretion following LPS stimulation [291][131]. Of note, BPs may also participate in the canonical antiviral response. Indeed, in response to IFN-γ and TNF-α, they showed upregulation of pro-IL-1β and pro-Caspase 1 mRNA expression [290][130]. Moreover, BPs express CCL2 after Poly:IC or LPS stimulation, relaying inflammatory signals from circulation to neurons, leading to elevated excitability [292][132].
Additionally, due to their location and the different route of entry for foreign agents (notably receptor-mediated endocytosis, unspecific transport by pinocytotic vesicles), BPs represent possible targets for viral infection. HIV infection can cause BBB disruption and contributes to the development of neurological dysfunction (e.g., HIV-associated neurological disorders—HAND). HIV is able to infect and replicate at low levels in BPs, using its co-receptors CXCR4 and CCR5 [222,293][37][133]. These cells even constitute one of HIV reservoirs in the central nervous system (CNS), the infection reactivation being possible thanks to genome integration mechanism. An increase in integrated genome in BPs is leading to a latent stage of infection following the initial peak of HIV production in the CNS [294,295][134][135]. Moreover, HIV infection of BPs has deleterious effects on BBB. Of note, it was shown that a decreased BPs coverage of BBB was a consequence of BPs dysfunction in HIV-infected patients [223][38], BPs coverage being negatively correlated with BBB permeability. The loss of pericytes observed has been associated with early PDGF-BB expression, which promotes pericytes migration away from their perivascular location [221][36]. Moreover, HIV infection leads to IL-6 secretion [222][37]. This may be another consequence of PDGF-BB secretion and downstream receptor signaling events [296][136]. IL-6 secretion by BPs also concurs in HIV-induced CNS damage and BBB disruption observed [222][37], the proinflammatory-induced response of endothelial cells causing BBB impairment [297][137].
HCMV infection may cause neurological pathologies either in children or immunocompromised patients. If, astrocytes and endothelial cells were initially reported as the main targets of HCMV at the BBB level [224][39], BPs were recently shown to be more permissive and sensitive to HCMV-induced lysis [225][40]. Thus, BPs are contributing to HCMV dissemination in CNS [30[40][77],225], as well as neuroinflammation via CXCL8, CXCL11, CCL5, TNF-α, IL-1β, and IL-6 secretion [225][40].
Japanese Encephalitis Virus (JEV) is a neurotropic mosquito-borne flavivirus that causes encephalitis, with neurological and psychological sequelae among a majority of survivors. Neuroinflammation is the most outstanding mechanism associated with JEV pathogenesis. BBB integrity is critical to regulate neuroinflammation, by limiting circulating immune cells entry and avoiding overwhelmed immune response. BPs are targeted by JEV as least in vitro. Following infection, they secrete IL-6 promoting proinflammatory responses and proteasomal degradation of Zonula Occludens-1 (ZO-1), thus participating in JEV-induced BBB impairment [226][41]. In addition, JEV was shown to trigger NF-κB through TLR7/MyD88 dependent axis, leading to PGE2 and CCL5/RANTES secretion [227][42]. The latter causes attraction and infiltration of leukocytes to the site of infection, provoking the aforementioned inappropriate immune response responsible for JEV neuropathology. The same mechanism is evoked for Herpes Simplex encephalitis but need to be further explored [228][43]. ZIKV, which is known for its neurological complications (e.g., microcephaly, Guillain-Barré Syndrome, and encephalitis) [230][45], can invade CNS through BPs infection at the level of the choroid plexus (via the receptor AXL) [229][44]. Both murine and human BPs are susceptible to ZIKV [229][44]. Furthermore, ZIKV-infected BPs are associated with BBB defect in vitro, illustrated by increased cytosolic ZO-1, decreased transepithelial electrical resistance and higher degree of FITC-dextran transport [229][44].
3. Osteoblasts and Viral Infections
Osteoblasts (OBs) are stromal cells, responsible for bone formation, through osteocalcin, ALP and type I collagen expression. As the other cells presented in this section, OBs (e.g., craniofacial bones) are derived from MSCs of the NC [298][138]. A great deal of pathogens are able to target bone tissues, leading to a variety of diseases ranging from caries to osteomyelitis. Among these pathogens, bacteria are the most cited. Yet the implication of viruses, and more specifically arthritogenic ones, in development of bone disorders should not be underestimated.
Ross River Virus (RRV) is a mosquito-borne alphavirus that generally causes flu-like illness and polyarthritis. As other alphaviruses, RRV might promote the development of bone diseases by targeting osteoblasts through Mxra8 [119][139]. Chen et al. addressed the effect of RRV infection on osteoblasts and showed that RRV-infected osteoblasts are producing high levels of IL-6 and CCL2 [233][48]. Additionally, this infection led to an imbalance in the Receptor Activator of Nuclear factor-KappaB Ligand (RANKL)/Osteoprotegerin (OPG) ratio in favor of osteoclastogenic activities and bone loss [233][48]. Moreover, the same team demonstrated an increased susceptibility to RRV in osteoblasts from osteoarthritic patients, further promoting the adverse effects of infection previously mentioned, due to a delayed IFN-β induction and RIG-I expression [232][47].
Similar outcomes are observed following OBs infection by Chikungunya virus (CHIKV), which is another arthritogenic alphavirus, best known for its recent epidemics between 2005 and 2006 in Reunion Island. Thus, an increased RANKL/OPG ratio in favor of osteoclastogenesis [234,299][49][140] can lead to bone loss through monocytic osteoclast recruitment [299][140].
HCV, besides its hepatic-related disease, might be able to trigger bone disorders. Indeed, HCV-infected patients have a higher risk of osteoporosis [300][141] and fracture [237][52]. Conversely, HCV infection was also associated with bone density hardening and osteosclerosis [235][50]. Once again, RANKL/OPG ratio impairment during HCV infection could be the cause of these detrimental consequences [235][50]. Kluger and colleagues addressed the permissiveness of OBs to HCV and provided evidence that OBs and osteoblast progenitors harbor HCV replication in the bone [236][51]. Thus, the implication of OBs in the pathophysiology of bone diseases following HCV infection should be explored given their involvement during alphaviruses-related disorders. On the other hand, as previously evoked, flavivirus ZIKV infection of BM-MSCs interferes with their ability to differentiate in osteoblasts, with a significant increase of IL-6 expression and a decrease of key osteoblast marker (e.g., ALP and RUNX2) in infected MSCs [197][12]. This susceptibility leads to impaired osteoblast function during ZIKV infection, triggering an imbalance in bone homeostasis and inducing bone-related disorders [197][12].
Measles morbillivirus (MeV), which belongs to the Paramyxoviridae family, is the cause of measles, a highly contagious disease. Symptoms of measles associate general symptoms (fever, cough) with a generalized maculopapular and erythematous rash. Moreover, MeV has been evoked in bone-related diseases such as otosclerosis [238,239][53][54] and Paget’s disease [240][55], both characterized by disturbed equilibrium of bone resorption and new bone formation. Several teams reported that productive OBs infection by MeV participates in the development of these disorders. Hence during MeV infection, OBs exhibit a higher expression of several osteogenic markers: bone morphogenic proteins (BMP-1, -4, -5, -6, and -7), ALP, bone sialo-protein, collagen 1α1 and OPG [238][53]. These findings highlight the ability of MeV to stimulate osteogenic differentiation of OBs thus participating in imbalance in bone formation. Such findings were corroborated by Potocka-Bakłażec et al., by demonstrating increased levels of TNF-α, IL-1β and decreased levels of OPG (conversely to Ayala-Peña et al. study) in MeV positive patients, also testifying bone remodeling subsequent to MeV infection [239][54].
HIV-infected patients have a higher incidence of osteopenia and osteoporosis due to bone demineralization and reduced bone mass. This is the result of OBs increased apoptosis following HIV infection in response both to expression of TNF-α and impaired Wnt/β-Catenin signaling in infected cells [231,301][46][142].
Osteoclasts, derived from macrophages, were thought to be the principal sensor of infection in the bone, due to their wide expression of PRRs. Notwithstanding, a growing body of evidence is in favor of PRRs’ expression by OBs, arguing for a sensing and antimicrobial role shared between osteoclasts and OBs [302][143]. Even if exacerbated inflammatory response in case of infection may lead to detrimental consequences, this has notably been exemplified earlier with IL-6 and development of arthritic pathologies, antiviral response remains essential to counteract most of virus-induced pathogenesis processes. Thus, the response of OBs in viral context has been addressed by Nakamura et al. on mouse osteoblastic cells (MC3T3-E1), using Poly:IC. They showed that in context of viral infection, OBs produce IFN-β as early as 1 h after stimulation due to Poly:IC recognition by TLR3, with a peak at 12 h [303][144]. Of note, they reported production of TLR3 and RIG-I in response to IFN-β, making OBs fully capable of viral dsRNA detection [303,304][144][145]. Of further note, IFN-β triggers CXCL10 production through a IFN-α/β receptor-STAT1 pathway [303][144]. Taken together these data suggest the ability of OBs to mount a proper IFN-type I response. Additionally, IL-27, a cytokine regulating immune responses as well as hematopoiesis and bone remodeling, was found to be expressed by OBs in inflammatory conditions (e.g., presence of type I IFN, IL-1β and TNF-α) [305][146]. So, OBs are thought to be part of a negative-feedback mechanism, limiting bone erosion and dampening T cell-mediated immune pathology during bone inflammation (therefore antiviral response).
4. Schwann Cell of Peripheral Nerves and Viral Infection
Schwann Cells (SCs) originate from NC. Moreover, MSCs are able to give rise to Schwann Cell-like cells after induction [306[147][148],307], these cells being able to drive a proper myelination. SCs are support cells that have a pivotal role in myelination of neurons from the peripheral nervous system (PNS). Thus, their affection is linked to demyelinating disorders of PNS (e.g., Guillain-Barré Syndrome).
As previously seen, HIV can cause neurological disorders, called HIV-associated neurological disorders (HAND). Among these HAND, wdistal ne canuropathy could be notably cite distal neuropathyd. Even if SCs are not primarily infected by HIV (infection has not been described in vitro), they express chemokine receptor CXCR4, a receptor for HIV-1 gp120 [241][56]. The pathway driven by HIV-1 gp120 leads to RANTES and TNF-α secretion, stimulating axon and neuron to release TNF receptor-1 (TNFR1) [241][56]. This promotes dorsal root ganglion neurotoxicity, including axon and myelin injury [241][56].
CNS tropism of Herpes Simplex Virus (HSV) and Herpes Zoster Virus/Varicella Zoster Virus (VZV) is well-established. Furthermore, infection of SCs by HSV or VZV was experimentally demonstrated [308,309][149][150]. However, it is difficult to say if this infection is of clinical relevance, since the principal mechanism evoked for HSV-induced GBS is a molecular mimicry of viral proteins, leading to cross-reacting antibodies [242][57]. However, this statement is based on case reports and needs to be further explored experimentally, in order to exclude any direct involvement of SCs in HSV-induced GBS.
Similarly, HCMV inclusions can be observed in SCs [243][58]. HCMV infection is the second most frequent infectious etiology of GBS [244][59], with notable cases in immunocompromised patients [245][60]. The pathogenesis of CMV-associated GBS have been linked to a probable molecular mimicry generating autoantibodies against, among others, moesin expressed by SCs [246][61]. Of note, these findings are called into question and need to be experimentally confirmed with an animal model, since no other team has obtained similar results [247][62].
As aforementioned, ZIKV is increasingly implicated in neurological disorders affecting CNS (i.e., microcephaly) as PNS (i.e., GBS) and of critical concern in the last years [230][45]. Initially, it has been demonstrated that CNS cells and oligodendrocytes, responsible for myelination in CNS, were more susceptible to ZIKV than PNS cells, causing axon and myelin injuries [248][63]. Yet more recent data indicate SCs are susceptible to ZIKV [310,311][151][152]. Volpi et al. showed that, in myelinating dorsal root ganglion explants from Ifnar1−/− mice, ZIKV infection of SCs leads to endoplasmic reticulum stress pathway activation and apoptosis in these cells, which finally cause demyelination and axon degeneration [310][151]. Additionally, Dhiman et al. showed a sustained viral production and a significant cell death at 96 h post-infection in vitro in human SCs, the infection inducing expression of proinflammatory cytokines (IL-6, TNF-α, IFN-β and IL-29) [311][152]. These works pave the way for a direct viral pathogenic effect or a cell-mediated inflammation in pathogenesis of ZIKV-associated GBS, even if a possible antibody dependent enhancement with Dengue Virus (DENV) sera or a demyelination induced cross-reactivity of anti-ZIKV antibodies are also discussed elsewhere [312,313][153][154].
Primary antiviral response mounted by SCs has not been well studied for each virus previously presented. However, SCs possess an efficient immune system to detect pathogens, represented by several PRRs. Among them, TLR3 and TLR7 are PRR devoted to virus detection and expressed by SCs [314][155]. TLR3 and TLR7 trigger a classical antiviral response after PAMP recognition. This response implies IFN response, driven by NF-κB and IRF, ISG release, and ultimately apoptosis, subsequently to inflammatory cytokine stimulation of extrinsic pathway.
References
- Yuan, Y.; Zhao, S.; Wang, X.; Teng, Z.; Li, D.; Zeng, Y. HIV-1 P55-Gag Protein Induces Senescence of Human Bone Marrow Mesenchymal Stem Cells and Reduces Their Capacity to Support Expansion of Hematopoietic Stem Cells in Vitro. Cell Biol. Int. 2017, 41, 969–981.
- Chandra, P.K.; Gerlach, S.L.; Wu, C.; Khurana, N.; Swientoniewski, L.T.; Abdel-Mageed, A.B.; Li, J.; Braun, S.E.; Mondal, D. Mesenchymal Stem Cells Are Attracted to Latent HIV-1-Infected Cells and Enable Virus Reactivation via a Non-Canonical PI3K-NFκB Signaling Pathway. Sci. Rep. 2018, 8, 14702.
- Cotter, E.J.; Chew, N.; Powderly, W.G.; Doran, P.P. HIV Type 1 Alters Mesenchymal Stem Cell Differentiation Potential and Cell Phenotype Ex Vivo. AIDS Res. Hum. Retrovir. 2010, 27, 187–199.
- Beaupere, C.; Garcia, M.; Larghero, J.; Fève, B.; Capeau, J.; Lagathu, C. The HIV Proteins Tat and Nef Promote Human Bone Marrow Mesenchymal Stem Cell Senescence and Alter Osteoblastic Differentiation. Aging Cell 2015, 14, 534–546.
- Sundin, M.; Örvell, C.; Rasmusson, I.; Sundberg, B.; Ringdén, O.; Le Blanc, K. Mesenchymal Stem Cells Are Susceptible to Human Herpesviruses, but Viral DNA Cannot Be Detected in the Healthy Seropositive Individual. Bone Marrow Transplant. 2006, 37, 1051–1059.
- Smirnov, S.V.; Harbacheuski, R.; Lewis-Antes, A.; Zhu, H.; Rameshwar, P.; Kotenko, S.V. Bone-Marrow-Derived Mesenchymal Stem Cells as a Target for Cytomegalovirus Infection: Implications for Hematopoiesis, Self-Renewal and Differentiation Potential. Virology 2007, 360, 6–16.
- Lee, M.-S.; Yuan, H.; Jeon, H.; Zhu, Y.; Yoo, S.; Shi, S.; Krueger, B.; Renne, R.; Lu, C.; Jung, J.U.; et al. Human Mesenchymal Stem Cells of Diverse Origins Support Persistent Infection with Kaposi’s Sarcoma-Associated Herpesvirus and Manifest Distinct Angiogenic, Invasive, and Transforming Phenotypes. mBio 2016, 7, e02109.
- Pessina, A.; Bonomi, A.; Coccè, V.; Bernardo, M.E.; Cometa, A.M.; Ferrari, M.; Sisto, F.; Cavicchini, L.; Locatelli, F. Assessment of Human Herpesvirus-6 Infection in Mesenchymal Stromal Cells Ex Vivo Expanded for Clinical Use. Transpl. Infect. Dis. Off. J. Transplant. Soc. 2009, 11, 491–496.
- Khatri, M.; Saif, Y.M. Influenza Virus Infects Bone Marrow Mesenchymal Stromal Cells in Vitro: Implications for Bone Marrow Transplantation. Cell Transplant. 2013, 22, 461–468.
- Thanunchai, M.; Kanrai, P.; Wiboon-Ut, S.; Puthavathana, P.; Hongeng, S.; Thitithanyanont, A. Tropism of Avian Influenza A (H5N1) Virus to Mesenchymal Stem Cells and CD34+ Hematopoietic Stem Cells. PLoS ONE 2013, 8, e81805.
- Cheung, M.B.; Sampayo-Escobar, V.; Green, R.; Moore, M.L.; Mohapatra, S.; Mohapatra, S.S. Respiratory Syncytial Virus-Infected Mesenchymal Stem Cells Regulate Immunity via Interferon Beta and Indoleamine-2,3-Dioxygenase. PLoS ONE 2016, 11, e0163709.
- Mumtaz, N.; Koedam, M.; van den Doel, P.B.; van Leeuwen, J.P.T.M.; Koopmans, M.P.G.; van der Eerden, B.C.J.; Rockx, B. Zika Virus Infection Perturbs Osteoblast Function. Sci. Rep. 2018, 8, 16975.
- Rong, Q.; Zhang, L.; Su, E.; Li, J.; Li, J.; Liu, Z.; Huang, Z.; Ma, W.; Cao, K.; Huang, J. Bone Marrow-Derived Mesenchymal Stem Cells Are Capable of Mediating Hepatitis B Virus Infection in Injured Tissues. J. Viral Hepat. 2008, 15, 607–614.
- Cardot-Leccia, N.; Hubiche, T.; Dellamonica, J.; Burel-Vandenbos, F.; Passeron, T. Pericyte Alteration Sheds Light on Micro-Vasculopathy in COVID-19 Infection. Intensive Care Med. 2020, 46, 1777–1778.
- Rendeiro, A.F.; Ravichandran, H.; Bram, Y.; Chandar, V.; Kim, J.; Meydan, C.; Park, J.; Foox, J.; Hether, T.; Warren, S.; et al. The Spatial Landscape of Lung Pathology during COVID-19 Progression. Nature 2021, 593, 564–569.
- Stephenson, S.E.; Wilson, C.L.; Bond, N.G.; Kaur, A.; Alvarez, X.; Midkiff, C.C.; Schnapp, L.M. Pericytes as Novel Targets for HIV/SIV Infection in the Lung. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2020, 319, L848–L853.
- Zhang, L.; Bansal, M.B. Role of Kupffer Cells in Driving Hepatic Inflammation and Fibrosis in HIV Infection. Front. Immunol. 2020, 11, 1086.
- Tuyama, A.C.; Hong, F.; Saiman, Y.; Wang, C.; Ozkok, D.; Mosoian, A.; Chen, P.; Chen, B.K.; Klotman, M.E.; Bansal, M.B. Human Immunodeficiency Virus (HIV)-1 Infects Human Hepatic Stellate Cells and Promotes Collagen I and Monocyte Chemoattractant Protein-1 Expression: Implications for the Pathogenesis of HIV/Hepatitis C Virus-Induced Liver Fibrosis. Hepatology 2010, 52, 612–622.
- Schulze-Krebs, A.; Preimel, D.; Popov, Y.; Bartenschlager, R.; Lohmann, V.; Pinzani, M.; Schuppan, D. Hepatitis C Virus-Replicating Hepatocytes Induce Fibrogenic Activation of Hepatic Stellate Cells. Gastroenterology 2005, 129, 246–258.
- Cheng, J.-C.; Tseng, C.-P.; Liao, M.-H.; Peng, C.-Y.; Yu, J.-S.; Chuang, P.-H.; Huang, J.-T.; Chen, J.J.W. Activation of Hepatic Stellate Cells by the Ubiquitin C-Terminal Hydrolase 1 Protein Secreted from Hepatitis C Virus-Infected Hepatocytes. Sci. Rep. 2017, 7, 4448.
- He, L.; Yuan, H.; Liang, J.; Hong, J.; Qu, C. Expression of Hepatic Stellate Cell Activation-Related Genes in HBV-, HCV-, and Nonalcoholic Fatty Liver Disease-Associated Fibrosis. PLoS ONE 2020, 15, e0233702.
- Salloum, S.; Holmes, J.A.; Jindal, R.; Bale, S.S.; Brisac, C.; Alatrakchi, N.; Lidofsky, A.; Kruger, A.J.; Fusco, D.N.; Luther, J.; et al. Exposure to Human Immunodeficiency Virus/Hepatitis C Virus in Hepatic and Stellate Cell Lines Reveals Cooperative Profibrotic Transcriptional Activation between Viruses and Cell Types: Salloum et Al. Hepatology 2016, 64, 1951–1968.
- Mazzocca, A.; Sciammetta, S.C.; Carloni, V.; Cosmi, L.; Annunziato, F.; Harada, T.; Abrignani, S.; Pinzani, M. Binding of Hepatitis C Virus Envelope Protein E2 to CD81 Up-Regulates Matrix Metalloproteinase-2 in Human Hepatic Stellate Cells. J. Biol. Chem. 2005, 280, 11329–11339.
- Bataller, R.; Paik, Y.; Lindquist, J.N.; Lemasters, J.J.; Brenner, D.A. Hepatitis C Virus Core and Nonstructural Proteins Induce Fibrogenic Effects in Hepatic Stellate Cells. Gastroenterology 2004, 126, 529–540.
- Wang, Q.; Zhou, J.; Zhang, B.; Tian, Z.; Tang, J.; Zheng, Y.; Huang, Z.; Tian, Y.; Jia, Z.; Tang, Y.; et al. Hepatitis B Virus Induces IL-23 Production in Antigen Presenting Cells and Causes Liver Damage via the IL-23/IL-17 Axis. PLoS Pathog. 2013, 9, e1003410.
- Tan, Z.; Qian, X.; Jiang, R.; Liu, Q.; Wang, Y.; Chen, C.; Wang, X.; Ryffel, B.; Sun, B. IL-17A Plays a Critical Role in the Pathogenesis of Liver Fibrosis through Hepatic Stellate Cell Activation. J. Immunol. 2013, 191, 1835–1844.
- Green, D.F.; Resnick, L.; Bourgoignie, J.J. HIV Infects Glomerular Endothelial and Mesangial but Not Epithelial Cells in Vitro. Kidney Int. 1992, 41, 956–960.
- Tokizawa, S.; Shimizu, N.; Hui-Yu, L.; Deyu, F.; Haraguchi, Y.; Oite, T.; Hoshino, H. Infection of Mesangial Cells with HIV and SIV: Identification of GPR1 as a Coreceptor. Kidney Int. 2000, 58, 607–617.
- Alcendor, D.J. Human Vascular Pericytes and Cytomegalovirus Pathobiology. Int. J. Mol. Sci. 2019, 20, 1456.
- Popik, W.; Correa, H.; Khatua, A.; Aronoff, D.M.; Alcendor, D.J. Mesangial Cells, Specialized Renal Pericytes and Cytomegalovirus Infectivity: Implications for HCMV Pathology in the Glomerular Vascular Unit and Post-Transplant Renal Disease. J. Transl. Sci. 2018, 5.
- Ustinov, J.A.; Loginov, R.J.; Mattila, P.M.; Nieminen, V.K.; Suni, J.I.; Häyry, P.J.; Lautenschlager, I.T. Cytomegalovirus Infection of Human Kidney Cells in Vitro. Kidney Int. 1991, 40, 954–960.
- Heieren, M.H.; van der Woude, F.J.; Balfour, H.H. Cytomegalovirus Replicates Efficiently in Human Kidney Mesangial Cells. Proc. Natl. Acad. Sci. USA 1988, 85, 1642–1646.
- Sansonno, D.; Gesualdo, L.; Manno, C.; Schena, F.P.; Dammacco, F. Hepatitis C Virus-Related Proteins in Kidney Tissue from Hepatitis C Virus-Infected Patients with Cryoglobulinemic Membranoproliferative Glomerulonephritis. Hepatology 1997, 25, 1237–1244.
- Alcendor, D.J. Zika Virus Infection of the Human Glomerular Cells: Implications for Viral Reservoirs and Renal Pathogenesis. J. Infect. Dis. 2017, 216, 162–171.
- Jansen, J.; Reimer, K.C.; Nagai, J.S.; Varghese, F.S.; Overheul, G.J.; de Beer, M.; Roverts, R.; Daviran, D.; Fermin, L.A.S.; Willemsen, B.; et al. SARS-CoV-2 Infects the Human Kidney and Drives Fibrosis in Kidney Organoids. Cell Stem Cell 2022, 29, 217–231.
- Niu, F.; Yao, H.; Zhang, W.; Sutliff, R.L.; Buch, S. Tat 101-Mediated Enhancement of Brain Pericyte Migration Involves Platelet-Derived Growth Factor Subunit B Homodimer: Implications for Human Immunodeficiency Virus-Associated Neurocognitive Disorders. J. Neurosci. 2014, 34, 11812–11825.
- Nakagawa, S.; Castro, V.; Toborek, M. Infection of Human Pericytes by HIV-1 Disrupts the Integrity of the Blood–Brain Barrier. J. Cell. Mol. Med. 2012, 16, 2950–2957.
- Persidsky, Y.; Hill, J.; Zhang, M.; Dykstra, H.; Winfield, M.; Reichenbach, N.L.; Potula, R.; Mukherjee, A.; Ramirez, S.H.; Rom, S. Dysfunction of Brain Pericytes in Chronic Neuroinflammation. J. Cereb. Blood Flow Metab. 2016, 36, 794–807.
- Cheeran, M.C.-J.; Lokensgard, J.R.; Schleiss, M.R. Neuropathogenesis of Congenital Cytomegalovirus Infection: Disease Mechanisms and Prospects for Intervention. Clin. Microbiol. Rev. 2009, 22, 99–126, Table of Contents.
- Alcendor, D.J.; Charest, A.M.; Zhu, W.Q.; Vigil, H.E.; Knobel, S.M. Infection and Upregulation of Proinflammatory Cytokines in Human Brain Vascular Pericytes by Human Cytomegalovirus. J. Neuroinflamm. 2012, 9, 95.
- Chen, C.-J.; Ou, Y.-C.; Li, J.-R.; Chang, C.-Y.; Pan, H.-C.; Lai, C.-Y.; Liao, S.-L.; Raung, S.-L.; Chang, C.-J. Infection of Pericytes In Vitro by Japanese Encephalitis Virus Disrupts the Integrity of the Endothelial Barrier. J. Virol. 2014, 88, 1150–1161.
- Chang, C.-Y.; Li, J.-R.; Ou, Y.-C.; Lin, S.-Y.; Wang, Y.-Y.; Chen, W.-Y.; Hu, Y.-H.; Lai, C.-Y.; Chang, C.-J.; Chen, C.-J. Interplay of Inflammatory Gene Expression in Pericytes Following Japanese Encephalitis Virus Infection. Brain Behav. Immun. 2017, 66, 230–243.
- Liu, H.; Qiu, K.; He, Q.; Lei, Q.; Lu, W. Mechanisms of Blood-Brain Barrier Disruption in Herpes Simplex Encephalitis. J. Neuroimmune Pharm. 2019, 14, 157–172.
- Kim, J.; Alejandro, B.; Hetman, M.; Hattab, E.M.; Joiner, J.; Schroten, H.; Ishikawa, H.; Chung, D.-H. Zika Virus Infects Pericytes in the Choroid Plexus and Enters the Central Nervous System through the Blood-Cerebrospinal Fluid Barrier. PLoS Pathog. 2020, 16, e1008204.
- Krauer, F.; Riesen, M.; Reveiz, L.; Oladapo, O.T.; Martínez-Vega, R.; Porgo, T.V.; Haefliger, A.; Broutet, N.J.; Low, N.; Group, W.Z.C.W. Zika Virus Infection as a Cause of Congenital Brain Abnormalities and Guillain–Barré Syndrome: Systematic Review. PLoS Med. 2017, 14, e1002203.
- Butler, J.S.; Dunning, E.C.; Murray, D.W.; Doran, P.P.; O’Byrne, J.M. HIV-1 Protein Induced Modulation of Primary Human Osteoblast Differentiation and Function via a Wnt/β-Catenin-Dependent Mechanism. J. Orthop. Res. 2013, 31, 218–226.
- Chen, W.; Foo, S.-S.; Li, R.W.; Smith, P.N.; Mahalingam, S. Osteoblasts from Osteoarthritis Patients Show Enhanced Susceptibility to Ross River Virus Infection Associated with Delayed Type I Interferon Responses. Virol. J. 2014, 11, 189.
- Chen, W.; Foo, S.-S.; Rulli, N.E.; Taylor, A.; Sheng, K.-C.; Herrero, L.J.; Herring, B.L.; Lidbury, B.A.; Li, R.W.; Walsh, N.C.; et al. Arthritogenic Alphaviral Infection Perturbs Osteoblast Function and Triggers Pathologic Bone Loss. Proc. Natl. Acad. Sci. USA 2014, 111, 6040–6045.
- Noret, M.; Herrero, L.; Rulli, N.; Rolph, M.; Smith, P.N.; Li, R.W.; Roques, P.; Gras, G.; Mahalingam, S. Interleukin 6, RANKL, and Osteoprotegerin Expression by Chikungunya Virus–Infected Human Osteoblasts. J. Infect. Dis. 2012, 206, 455–457.
- Manganelli, P.; Giuliani, N.; Fietta, P.; Mancini, C.; Lazzaretti, M.; Pollini, A.; Quaini, F.; Pedrazzoni, M. OPG/RANKL System Imbalance in a Case of Hepatitis C-Associated Osteosclerosis: The Pathogenetic Key? Clin. Rheumatol. 2005, 24, 296–300.
- Kluger, R.; Mühlberger, H.; Hoffmann, O.; Berger, C.E.; Engel, A.; Pavlova, B.G. Osteoprogenitor Cells and Osteoblasts Are Targets for Hepatitis C Virus. Clin. Orthop. Relat. Res. 2005, 433, 251–257.
- Hansen, A.-B.E.; Omland, L.H.; Krarup, H.; Obel, N. Fracture Risk in Hepatitis C Virus Infected Persons: Results from the DANVIR Cohort Study. J. Hepatol. 2014, 61, 15–21.
- Ayala-Peña, V.; Santillán, G.; Scolaro, L. Experimental in Vitro Infection of Rat Osteoblasts with Measles Virus Stimulates Osteogenic Differentiation. Biochem. Biophys. Res. Commun. 2014, 451, 609–614.
- Potocka-Bakłażec, M.; Sakowicz-Burkiewicz, M.; Kuczkowski, J.; Pawełczyk, T.; Stankiewicz, C.; Sierszeń, W.; Jankowski, Z.; Buczny, J. Expression of TNF-α, OPG, IL-1β and the Presence of the Measles Virus RNA in the Stapes of the Patients with Otosclerosis. Eur. Arch. Oto-Rhino-Laryngol. 2015, 272, 1907–1912.
- Teramachi, J.; Nagata, Y.; Mohammad, K.; Inagaki, Y.; Ohata, Y.; Guise, T.; Michou, L.; Brown, J.P.; Windle, J.J.; Kurihara, N.; et al. Measles Virus Nucleocapsid Protein Increases Osteoblast Differentiation in Paget’s Disease. J. Clin. Investig. 2016, 126, 1012–1022.
- Keswani, S.C.; Polley, M.; Pardo, C.A.; Griffin, J.W.; McArthur, J.C.; Hoke, A. Schwann Cell Chemokine Receptors Mediate HIV-1 Gp120 Toxicity to Sensory Neurons. Ann. Neurol. 2003, 54, 287–296.
- Dilena, R.; Strazzer, S.; Esposito, S.; Paglialonga, F.; Tadini, L.; Barbieri, S.; Giannini, A. Locked-in–like Fulminant Infantile Guillain–Barré Syndrome Associated with Herpes Simplex Virus 1 Infection. Muscle Nerve 2016, 53, 140–143.
- Behar, R.; Wiley, C.; McCutchan, J.A. Cytomegalovirus Polyradiculoneuropathy in Acquired Immune Deficiency Syndrome. Neurology 1987, 37, 557.
- Orlikowski, D.; Porcher, R.; Sivadon-Tardy, V.; Quincampoix, J.-C.; Raphael, J.-C.; Durand, M.-C.; Sharshar, T.; Roussi, J.; Caudie, C.; Annane, D.; et al. Guillain-Barre Syndrome Following Primary Cytomegalovirus Infection: A Prospective Cohort Study. Clin. Infect. Dis. 2011, 52, 837–844.
- Mohan, A.; Smith-Rohrberg, D.; Sethu, M.; Sharma, S.K. Cytomegalovirus Polyradiculopathy: A Rare Neurological Manifestation of Acquired Immunodeficiency Syndrome. Neurol. India 2008, 56, 493–494.
- Sawai, S.; Satoh, M.; Mori, M.; Misawa, S.; Sogawa, K.; Kazami, T.; Ishibashi, M.; Beppu, M.; Shibuya, K.; Ishige, T.; et al. Moesin Is a Possible Target Molecule for Cytomegalovirus-Related Guillain-Barre Syndrome. Neurology 2014, 83, 113–117.
- Miyaji, K.; Devaux, J.; Yuki, N.; Sawai, S.; Mori, M.; Kuwabara, S.; Miyaji, K.; Yuki, N.; Sawai, S.; Mori, M.; et al. Moesin Is a Possible Target Molecule for Cytomegalovirus-Related Guillain-Barre Syndrome. Neurology 2014, 83, 2314–2315.
- Cumberworth, S.L.; Barrie, J.A.; Cunningham, M.E.; de Figueiredo, D.P.G.; Schultz, V.; Wilder-Smith, A.J.; Brennan, B.; Pena, L.J.; Freitas de Oliveira França, R.; Linington, C.; et al. Zika Virus Tropism and Interactions in Myelinating Neural Cell Cultures: CNS Cells and Myelin Are Preferentially Affected. Acta Neuropathol. Commun. 2017, 5, 50.
- Caplan, A.I. New MSC: MSCs as Pericytes Are Sentinels and Gatekeepers. J. Orthop. Res. 2017, 35, 1151–1159.
- Le Blanc, K.; Davies, L.C. Mesenchymal Stromal Cells and the Innate Immune Response. Immunol. Lett. 2015, 168, 140–146.
- Bernardo, M.E.; Fibbe, W.E. Mesenchymal Stromal Cells: Sensors and Switchers of Inflammation. Cell Stem Cell 2013, 13, 392–402.
- Waterman, R.S.; Tomchuck, S.L.; Henkle, S.L.; Betancourt, A.M. A New Mesenchymal Stem Cell (MSC) Paradigm: Polarization into a Pro-Inflammatory MSC1 or an Immunosuppressive MSC2 Phenotype. PLoS ONE 2010, 5, e10088.
- Tomchuck, S.L.; Zwezdaryk, K.J.; Coffelt, S.B.; Waterman, R.S.; Danka, E.S.; Scandurro, A.B. Toll-like Receptors on Human Mesenchymal Stem Cells Drive Their Migration and Immunomodulating Responses. Stem Cells 2008, 26, 99–107.
- Friedenstein, A.J.; Chailakhyan, R.K.; Latsinik, N.V.; Panasyuk, A.F.; Keiliss-Borok, I.V. Stromal Cells Responsible for Transferring the Microenvironment of the Hemopoietic Tissues. Cloning in Vitro and Retransplantation in Vivo. Transplantation 1974, 17, 331–340.
- Isern, J.; Garcia-Garcia, A.; Martin, A.M.; Arranz, L.; Martin-Perez, D.; Torroja, C.; Sanchez-Csabo, F.; Mendez-Ferrer, S. The Neural Crest Is a Source of Mesenchymal Stem Cells with Specialized Hematopoietic Stem-Cell-Niche Function. eLife 2014, 3, e03696.
- Mendez-Ferrer, S.; Michurina, T.V.; Ferraro, F.; Mazloom, A.R.; MacArthur, B.D.; Lira, S.A.; Scadden, D.T.; Ma’ayan, A.; Enikolopov, G.N.; Frenette, P.S. Mesenchymal and Haematopoietic Stem Cells Form a Unique Bone Marrow Niche. Nature 2010, 466, 829–834.
- Schneider, R.K.; Mullally, A.; Dugourd, A.; Peisker, F.; Hoogenboezem, R.; Van Strien, P.M.H.; Bindels, E.M.; Heckl, D.; Büsche, G.; Fleck, D.; et al. Gli1+ Mesenchymal Stromal Cells Are a Key Driver of Bone Marrow Fibrosis and an Important Cellular Therapeutic Target. Cell Stem Cell 2017, 20, 785–800.
- Sacchetti, B.; Funari, A.; Michienzi, S.; Di Cesare, S.; Piersanti, S.; Saggio, I.; Tagliafico, E.; Ferrari, S.; Robey, P.G.; Riminucci, M.; et al. Self-Renewing Osteoprogenitors in Bone Marrow Sinusoids Can Organize a Hematopoietic Microenvironment. Cell 2007, 131, 324–336.
- Hmadcha, A.; Martin-Montalvo, A.; Gauthier, B.R.; Soria, B.; Capilla-Gonzalez, V. Therapeutic Potential of Mesenchymal Stem Cells for Cancer Therapy. Front. Bioeng. Biotechnol. 2020, 8, 43.
- Collino, F.; Bruno, S.; Incarnato, D.; Dettori, D.; Neri, F.; Provero, P.; Pomatto, M.; Oliviero, S.; Tetta, C.; Quesenberry, P.J.; et al. AKI Recovery Induced by Mesenchymal Stromal Cell-Derived Extracellular Vesicles Carrying MicroRNAs. J. Am. Soc. Nephrol. 2015, 26, 2349–2360.
- Kallmeyer, K.; Ryder, M.A.; Pepper, M.S. Mesenchymal Stromal Cells: A Possible Reservoir for HIV-1? Stem Cell Rev. Rep. 2022, 18, 1253–1280.
- Soland, M.A.; Keyes, L.R.; Bayne, R.; Moon, J.; Porada, C.D.; Jeor, S.S.; Almeida-Porada, G. Perivascular Stromal Cells as a Potential Reservoir of Human Cytomegalovirus. Am. J. Transplant. 2014, 14, 820–830.
- Rollín, R.; Álvarez-Lafuente, R.; Marco, F.; Jover, J.A.; Hernández-García, C.; Rodríguez-Navas, C.; López-Durán, L.; Fernández-Gutiérrez, B. Human Parvovirus B19, Varicella Zoster Virus, and Human Herpesvirus-6 in Mesenchymal Stem Cells of Patients with Osteoarthritis: Analysis with Quantitative Real-Time Polymerase Chain Reaction. Osteoarthr. Cartil. 2007, 15, 475–478.
- Le Blanc, K.; Mougiakakos, D. Multipotent Mesenchymal Stromal Cells and the Innate Immune System. Nat. Rev. Immunol. 2012, 12, 383–396.
- Raicevic, G.; Najar, M.; Busser, H.; Crompot, E.; Bron, D.; Toungouz, M.; Lagneaux, L. Comparison and Immunobiological Characterization of Retinoic Acid Inducible Gene-I-like Receptor Expression in Mesenchymal Stromal Cells. Sci. Rep. 2017, 7, 2896.
- Mastri, M.; Shah, Z.; McLaughlin, T.; Greene, C.J.; Baum, L.; Suzuki, G.; Lee, T. Activation of Toll-like Receptor 3 Amplifies Mesenchymal Stem Cell Trophic Factors and Enhances Therapeutic Potency. Am. J. Physiol.-Cell Physiol. 2012, 303, C1021–C1033.
- Raicevic, G.; Rouas, R.; Najar, M.; Stordeur, P.; Id Boufker, H.; Bron, D.; Martiat, P.; Goldman, M.; Nevessignsky, M.T.; Lagneaux, L. Inflammation Modifies the Pattern and the Function of Toll-like Receptors Expressed by Human Mesenchymal Stromal Cells. Hum. Immunol. 2010, 71, 235–244.
- Meisel, R.; Brockers, S.; Heseler, K.; Degistirici, Ö.; Bülle, H.; Woite, C.; Stuhlsatz, S.; Schwippert, W.; Jäger, M.; Sorg, R.; et al. Human but Not Murine Multipotent Mesenchymal Stromal Cells Exhibit Broad-Spectrum Antimicrobial Effector Function Mediated by Indoleamine 2,3-Dioxygenase. Leukemia 2011, 25, 648–654.
- Sengupta, V.; Sengupta, S.; Lazo, A.; Woods, P.; Nolan, A.; Bremer, N. Exosomes Derived from Bone Marrow Mesenchymal Stem Cells as Treatment for Severe COVID-19. Stem Cells Dev. 2020, 29, 747–754.
- Sabatini, F.; Petecchia, L.; Tavian, M.; de Villeroché, V.J.; Rossi, G.A.; Brouty-Boyé, D. Human Bronchial Fibroblasts Exhibit a Mesenchymal Stem Cell Phenotype and Multilineage Differentiating Potentialities. Lab. Investig. 2005, 85, 962–971.
- Sinclair, K.; Yerkovich, S.T.; Chambers, D.C. Mesenchymal Stem Cells and the Lung. Respirology 2013, 18, 397–411.
- Humphreys, B.D.; Lin, S.-L.; Kobayashi, A.; Hudson, T.E.; Nowlin, B.T.; Bonventre, J.V.; Valerius, M.T.; McMahon, A.P.; Duffield, J.S. Fate Tracing Reveals the Pericyte and Not Epithelial Origin of Myofibroblasts in Kidney Fibrosis. Am. J. Pathol. 2010, 176, 85–97.
- Kramann, R.; Schneider, R.K.; DiRocco, D.P.; Machado, F.; Fleig, S.; Bondzie, P.A.; Henderson, J.M.; Ebert, B.L.; Humphreys, B.D. Perivascular Gli1+ Progenitors Are Key Contributors to Injury-Induced Organ Fibrosis. Cell Stem Cell 2015, 16, 51–66.
- Avanzini, M.A.; Mura, M.; Percivalle, E.; Bastaroli, F.; Croce, S.; Valsecchi, C.; Lenta, E.; Nykjaer, G.; Cassaniti, I.; Bagnarino, J.; et al. Human Mesenchymal Stromal Cells Do Not Express ACE2 and TMPRSS2 and Are Not Permissive to SARS-CoV-2 Infection. Stem Cells Transl. Med. 2021, 10, 636–642.
- Zuk, P.A.; Zhu, M.; Ashjian, P.; De Ugarte, D.A.; Huang, J.I.; Mizuno, H.; Alfonso, Z.C.; Fraser, J.K.; Benhaim, P.; Hedrick, M.H. Human Adipose Tissue Is a Source of Multipotent Stem Cells. MBoC 2002, 13, 4279–4295.
- Uzbas, F.; May, I.D.; Parisi, A.M.; Thompson, S.K.; Kaya, A.; Perkins, A.D.; Memili, E. Molecular Physiognomies and Applications of Adipose-Derived Stem Cells. Stem Cell Rev. Rep. 2015, 11, 298–308.
- Bajek, A.; Gurtowska, N.; Olkowska, J.; Kazmierski, L.; Maj, M.; Drewa, T. Adipose-Derived Stem Cells as a Tool in Cell-Based Therapies. Arch. Immunol. Ther. Exp. 2016, 64, 443–454.
- Fang, Y.; Zhang, Y.; Zhou, J.; Cao, K. Adipose-Derived Mesenchymal Stem Cell Exosomes: A Novel Pathway for Tissues Repair. Cell Tissue Bank 2019, 20, 153–161.
- Kingham, P.J.; Kalbermatten, D.F.; Mahay, D.; Armstrong, S.J.; Wiberg, M.; Terenghi, G. Adipose-Derived Stem Cells Differentiate into a Schwann Cell Phenotype and Promote Neurite Outgrowth in Vitro. Exp. Neurol. 2007, 207, 267–274.
- Ouchi, N.; Parker, J.L.; Lugus, J.J.; Walsh, K. Adipokines in Inflammation and Metabolic Disease. Nat. Rev. Immunol. 2011, 11, 85–97.
- Vernochet, C.; Azoulay, S.; Duval, D.; Guedj, R.; Cottrez, F.; Vidal, H.; Ailhaud, G.; Dani, C. Human Immunodeficiency Virus Protease Inhibitors Accumulate into Cultured Human Adipocytes and Alter Expression of Adipocytokines. J. Biol. Chem. 2005, 280, 2238–2243.
- Wang, Y.; Wang, F.; Zhao, H.; Zhang, X.; Chen, H.; Zhang, K. Human Adipose-Derived Mesenchymal Stem Cells Are Resistant to HBV Infection during Differentiation into Hepatocytes in Vitro. Int. J. Mol. Sci. 2014, 15, 6096–6110.
- Choi, J.E.; Hur, W.; Kim, J.-H.; Li, T.Z.; Lee, E.B.; Lee, S.W.; Kang, W.; Shin, E.-C.; Wakita, T.; Yoon, S.K. MicroRNA-27a Modulates HCV Infection in Differentiated Hepatocyte-Like Cells from Adipose Tissue-Derived Mesenchymal Stem Cells. PLoS ONE 2014, 9, e91958.
- Geerts, A. History, Heterogeneity, Developmental Biology, and Functions of Quiescent Hepatic Stellate Cells. Semin. Liver Dis. 2001, 21, 311–336.
- Kupffer, C. Ueber Sternzellen der Leber: Briefliche Mittheilung an Prof. Waldeyer. Arch. Mikrosk. Anat. 1876, 12, 353–358.
- Cassiman, D.; Libbrecht, L.; Desmet, V.; Denef, C.; Roskams, T. Hepatic Stellate Cell/Myofibroblast Subpopulations in Fibrotic Human and Rat Livers. J. Hepatol. 2002, 36, 200–209.
- Neubauer, K.; Knittel, T.; Aurisch, S.; Fellmer, P.; Ramadori, G. Glial Fibrillary Acidic Protein—A Cell Type Specific Marker for Ito Cells in Vivo and in Vitro. J. Hepatol. 1996, 24, 719–730.
- Ito, T.; Nemoto, M. Über Die Kupfferschen Sternzellen Und Die “Fettspeicherungszellen” (“Fat Storing Cells”) in Der Blutkapillarenwand Der Menschlichen Leber. Okajimas Folia Anat. Jpn. 1952, 24, 243–258.
- Asada, N.; Takase, M.; Nakamura, J.; Oguchi, A.; Asada, M.; Suzuki, N.; Yamarnura, K.; Nagoshi, N.; Shibata, S.; Rao, T.N.; et al. Dysfunction of Fibroblasts of Extrarenal Origin Underlies Renal Fibrosis and Renal Anemia in Mice. J. Clin. Investig. 2011, 121, 3981–3990.
- Miyata, E.; Masuya, M.; Yoshida, S.; Nakamura, S.; Kato, K.; Sugimoto, Y.; Shibasaki, T.; Yamamura, K.; Ohishi, K.; Nishii, K.; et al. Hematopoietic Origin of Hepatic Stellate Cells in the Adult Liver. Blood 2008, 111, 2427–2435.
- Friedman, S.L. Hepatic Stellate Cells: Protean, Multifunctional, and Enigmatic Cells of the Liver. Physiol. Rev. 2008, 88, 125–172.
- Wake, K. Perisinusoidal Stellate Cells (Fat-Storing Cells, Interstitial Cells, Lipocytes), Their Related Structure in and around the Liver Sinusoids, and Vitamin A-Storing Cells in Extrahepatic Organs. In International Review of Cytology; Elsevier: Amsterdam, The Netherlands, 1980; Volume 66, pp. 303–353. ISBN 978-0-12-364466-4.
- Zhang, C.-Y.; Yuan, W.-G.; He, P.; Lei, J.-H.; Wang, C.-X. Liver Fibrosis and Hepatic Stellate Cells: Etiology, Pathological Hallmarks and Therapeutic Targets. World J. Gastroenterol. 2016, 22, 10512.
- Yan, C.; Zhou, L.; Han, Y.-P. Contribution of Hepatic Stellate Cells and Matrix Metalloproteinase 9 in Acute Liver Failure: HSCs and MMP 9 in ALF. Liver Int. 2008, 28, 959–971.
- Gupta, G.; Khadem, F.; Uzonna, J.E. Role of Hepatic Stellate Cell (HSC)-Derived Cytokines in Hepatic Inflammation and Immunity. Cytokine 2019, 124, 154542.
- Wang, B.; Trippler, M.; Pei, R.; Lu, M.; Broering, R.; Gerken, G.; Schlaak, J.F. Toll-like Receptor Activated Human and Murine Hepatic Stellate Cells Are Potent Regulators of Hepatitis C Virus Replication. J. Hepatol. 2009, 51, 1037–1045.
- Da Silva Meirelles, L.; Caplan, A.I.; Nardi, N.B. In Search of the In Vivo Identity of Mesenchymal Stem Cells. Stem Cells 2008, 26, 2287–2299.
- Lin, S.-L.; Kisseleva, T.; Brenner, D.A.; Duffield, J.S. Pericytes and Perivascular Fibroblasts Are the Primary Source of Collagen-Producing Cells in Obstructive Fibrosis of the Kidney. Am. J. Pathol. 2008, 173, 1617–1627.
- Duffield, J.S. Cellular and Molecular Mechanisms in Kidney Fibrosis. J. Clin. Investig. 2014, 124, 2299–2306.
- Blüm, P.; Pircher, J.; Merkle, M.; Czermak, T.; Ribeiro, A.; Mannell, H.; Krötz, F.; Hennrich, A.; Spannagl, M.; Köppel, S.; et al. Arterial Thrombosis in the Context of HCV-Associated Vascular Disease Can Be Prevented by Protein C. Cell. Mol. Immunol. 2017, 14, 986–996.
- Kobayashi, N.; Bagheri, N.; Nedrud, J.G.; Strieter, R.M.; Tomino, Y.; Lamm, M.E.; Emancipator, S.N. Differential Effects of Sendai Virus Infection on Mediator Synthesis by Mesangial Cells from Two Mouse Strains. Kidney Int. 2003, 64, 1675–1684.
- Pasch, A.; Frey, F.J. Coxsackie B Viruses and the Kidney—A Neglected Topic. Nephrol. Dial. Transplant. 2006, 21, 1184–1187.
- Popik, W.; Khatua, A.K.; Fabre, N.F.; Hildreth, J.E.K.; Alcendor, D.J. BK Virus Replication in the Glomerular Vascular Unit: Implications for BK Virus Associated Nephropathy. Viruses 2019, 11, 583.
- Zhai, S.; Hu, L.; Zhong, L.; Guo, Y.; Dong, L.; Jia, R.; Wang, Z. Respiratory Syncytial Virus Aggravates Renal Injury through Cytokines and Direct Renal Injury. Front. Cell. Infect. Microbiol. 2016, 6, 112.
- Mattana, J.; Abramovici, M.; Singhal, P.C. Effects of Human Immunodeficiency Virus Sera and Macrophage Supernatants on Mesangial Cell Proliferation and Matrix Synthesis. Am. J. Pathol. 1993, 143, 814–822.
- Kotton, C.N.; Fishman, J.A. Viral Infection in the Renal Transplant Recipient. J. Am. Soc. Nephrol. 2005, 16, 1758–1774.
- Baumert, T.F.; Berg, T.; Lim, J.K.; Nelson, D.R. Status of Direct-Acting Antiviral Therapy for Hepatitis C Virus Infection and Remaining Challenges. Gastroenterology 2019, 156, 431–445.
- Flür, K.; Allam, R.; Zecher, D.; Kulkarni, O.P.; Lichtnekert, J.; Schwarz, M.; Beutler, B.; Vielhauer, V.; Anders, H.-J. Viral RNA Induces Type I Interferon-Dependent Cytokine Release and Cell Death in Mesangial Cells via Melanoma-Differentiation-Associated Gene-5: Implications for Viral Infection-Associated Glomerulonephritis. Am. J. Pathol. 2009, 175, 2014–2022.
- Imaizumi, T.; Tanaka, H.; Matsumiya, T.; Yoshida, H.; Tanji, K.; Tsuruga, K.; Oki, E.; Aizawa-Yashiro, T.; Ito, E.; Satoh, K. Retinoic Acid-Inducible Gene-I Is Induced by Double-Stranded RNA and Regulates the Expression of CC Chemokine Ligand (CCL) 5 in Human Mesangial Cells. Nephrol. Dial. Transplant. 2010, 25, 3534–3539.
- Allam, R.; Lichtnekert, J.; Moll, A.G.; Taubitz, A.; Vielhauer, V.; Anders, H.-J. Viral RNA and DNA Trigger Common Antiviral Responses in Mesangial Cells. J. Am. Soc. Nephrol. 2009, 20, 1986–1996.
- Patole, P.S.; Gröne, H.-J.; Segerer, S.; Ciubar, R.; Belemezova, E.; Henger, A.; Kretzler, M.; Schlöndorff, D.; Anders, H.-J. Viral Double-Stranded RNA Aggravates Lupus Nephritis through Toll-Like Receptor 3 on Glomerular Mesangial Cells and Antigen-Presenting Cells. J. Am. Soc. Nephrol. 2005, 16, 1326–1338.
- Appaix, F. Brain Mesenchymal Stem Cells: The Other Stem Cells of the Brain? World J. Stem Cells 2014, 6, 134–143.
- Armulik, A.; Genove, G.; Betsholtz, C. Pericytes: Developmental, Physiological, and Pathological Perspectives, Problems, and Promises. Dev. Cell 2011, 21, 193–215.
- Dupin, E.; Sommer, L. Neural Crest Progenitors and Stem Cells: From Early Development to Adulthood. Dev. Biol. 2012, 366, 83–95.
- Nyúl-Tóth, Á.; Kozma, M.; Nagyőszi, P.; Nagy, K.; Fazakas, C.; Haskó, J.; Molnár, K.; Farkas, A.E.; Végh, A.G.; Váró, G.; et al. Expression of Pattern Recognition Receptors and Activation of the Non-Canonical Inflammasome Pathway in Brain Pericytes. Brain Behav. Immun. 2017, 64, 220–231.
- Kovac, A.; Erickson, M.A.; Banks, W.A. Brain Microvascular Pericytes Are Immunoactive in Culture: Cytokine, Chemokine, Nitric Oxide, and LRP-1 Expression in Response to Lipopolysaccharide. J. Neuroinflamm. 2011, 8, 139.
- Duan, L.; Zhang, X.-D.; Miao, W.-Y.; Sun, Y.-J.; Xiong, G.; Wu, Q.; Li, G.; Yang, P.; Yu, H.; Li, H.; et al. PDGFRβ Cells Rapidly Relay Inflammatory Signal from the Circulatory System to Neurons via Chemokine CCL2. Neuron 2018, 100, 183–200.e8.
- Cho, H.J.; Kuo, A.M.-S.; Bertrand, L.; Toborek, M. HIV Alters Gap Junction-Mediated Intercellular Communication in Human Brain Pericytes. Front. Mol. Neurosci. 2017, 10, 410.
- Bertrand, L.; Cho, H.J.; Toborek, M. Blood–Brain Barrier Pericytes as a Target for HIV-1 Infection. Brain 2019, 142, 502–511.
- Piekna-Przybylska, D.; Nagumotu, K.; Reid, D.M.; Maggirwar, S.B. HIV-1 Infection Renders Brain Vascular Pericytes Susceptible to the Extracellular Glutamate. J. Neurovirol. 2019, 25, 114–126.
- Gaceb, A.; Özen, I.; Padel, T.; Barbariga, M.; Paul, G. Pericytes Secrete Pro-Regenerative Molecules in Response to Platelet-Derived Growth Factor-BB. J. Cereb. Blood Flow Metab. 2018, 38, 45–57.
- Blecharz-Lang, K.G.; Wagner, J.; Fries, A.; Nieminen-Kelhä, M.; Rösner, J.; Schneider, U.C.; Vajkoczy, P. Interleukin 6-Mediated Endothelial Barrier Disturbances Can Be Attenuated by Blockade of the IL6 Receptor Expressed in Brain Microvascular Endothelial Cells. Transl. Stroke Res. 2018, 9, 631–642.
- Saint-Pastou Terrier, C.; Gasque, P. Bone Responses in Health and Infectious Diseases: A Focus on Osteoblasts. J. Infect. 2017, 75, 281–292.
- Zhang, Q.; Fu, L.; Liang, Y.; Guo, Z.; Wang, L.; Ma, C.; Wang, H. Exosomes Originating from MSCs Stimulated with TGF-β and IFN-γ Promote Treg Differentiation. J. Cell. Physiol. 2018, 233, 6832–6840.
- Chen, W.; Foo, S.-S.; Taylor, A.; Lulla, A.; Merits, A.; Hueston, L.; Forwood, M.R.; Walsh, N.C.; Sims, N.A.; Herrero, L.J.; et al. Bindarit, an Inhibitor of Monocyte Chemotactic Protein Synthesis, Protects against Bone Loss Induced by Chikungunya Virus Infection. J. Virol. 2015, 89, 581–593.
- Chen, C.-H.; Lin, C.-L.; Kao, C.-H. Relation Between Hepatitis C Virus Exposure and Risk of Osteoporosis: A Nationwide Population-Based Study. Medicine 2015, 94, e2086.
- Gibellini, D.; Crignis, E.D.; Ponti, C.; Cimatti, L.; Borderi, M.; Tschon, M.; Giardino, R.; Re, M.C. HIV-1 Triggers Apoptosis in Primary Osteoblasts and HOBIT Cells through TNFα Activation. J. Med. Virol. 2008, 80, 1507–1514.
- Bar-Shavit, Z. Taking a Toll on the Bones: Regulation of Bone Metabolism by Innate Immune Regulators. Autoimmunity 2008, 41, 195–203.
- Nakamura, K.; Deyama, Y.; Yoshimura, Y.; Suzuki, K.; Morita, M. Toll-like Receptor 3 Ligand-Induced Antiviral Response in Mouse Osteoblastic Cells. Int. J. Mol. Med. 2007, 19, 771–775.
- Nakamura, K.; Deyama, Y.; Yoshimura, Y.; Suzuki, K.; Morita, M. Synthetic Double-Stranded RNA Induces Retinoic Acid-Inducible Gene-I in Mouse Osteoblastic Cells. Mol. Med. Rep. 2008, 1, 833–836.
- Larousserie, F.; Bsiri, L.; Dumaine, V.; Dietrich, C.; Audebourg, A.; Radenen-Bussière, B.; Anract, P.; Vacher-Lavenu, M.-C.; Devergne, O. Frontline Science: Human Bone Cells as a Source of IL-27 under Inflammatory Conditions: Role of TLRs and Cytokines. J. Leukoc. Biol. 2017, 101, 1289–1300.
- Matsuse, D.; Kitada, M.; Kohama, M.; Nishikawa, K.; Makinoshima, H.; Wakao, S.; Fujiyoshi, Y.; Heike, T.; Nakahata, T.; Akutsu, H.; et al. Human Umbilical Cord-Derived Mesenchymal Stromal Cells Differentiate Into Functional Schwann Cells That Sustain Peripheral Nerve Regeneration. J. Neuropathol. Exp. Neurol. 2010, 69, 973–985.
- Sun, X.; Zhu, Y.; Yin, H.; Guo, Z.; Xu, F.; Xiao, B.; Jiang, W.; Guo, W.; Meng, H.; Lu, S.; et al. Differentiation of Adipose-Derived Stem Cells into Schwann Cell-like Cells through Intermittent Induction: Potential Advantage of Cellular Transient Memory Function. Stem Cell Res. Ther. 2018, 9, 133.
- Assouline, J.G.; Levin, M.J.; Straus, E.; Ostrovet, J.M. Varicella-Zoster Virus Infection of Human Astrocytes, Schwann Ceils, and Neurons. Virology 1990, 179, 834–844.
- Shimeld, C.; Efstathiou, S.; Hill, T. Tracking the Spread of a LacZ-Tagged Herpes Simplex Virus Type 1 between the Eye and the Nervous System of the Mouse: Comparison of Primary and Recurrent Infection. J. Virol. 2001, 75, 5252–5262.
- Volpi, V.G.; Pagani, I.; Ghezzi, S.; Iannacone, M.; D’Antonio, M.; Vicenzi, E. Zika Virus Replication in Dorsal Root Ganglia Explants from Interferon Receptor1 Knockout Mice Causes Myelin Degeneration. Sci. Rep. 2018, 8, 10166.
- Dhiman, G.; Abraham, R.; Griffin, D.E. Human Schwann Cells Are Susceptible to Infection with Zika and Yellow Fever Viruses, but Not Dengue Virus. Sci. Rep. 2019, 9, 9951.
- Muñoz, L.; Barreras, P.; Pardo, C. Zika Virus–Associated Neurological Disease in the Adult: Guillain–Barré Syndrome, Encephalitis, and Myelitis. Semin. Reprod. Med. 2016, 34, 273–279.
- Lebeau, G.; Frumence, E.; Turpin, J.; Begue, F.; Hoarau, J.-J.; Gadea, G.; Krejbich-Trotot, P.; Desprès, P.; Viranaicken, W. Zika E Glycan Loop Region and Guillain–Barré Syndrome-Related Proteins: A Possible Molecular Mimicry to Be Taken in Account for Vaccine Development. Vaccines 2021, 9, 283.
- Goethals, S.; Ydens, E.; Timmerman, V.; Janssens, S. Toll-like Receptor Expression in the Peripheral Nerve. Glia 2010, 58, 1701–1709.
More