Hsp90 is a molecular chaperone with over 300 client proteins that act in the cell cycle and in signalling processes. Disruption of Hsp90 chaperone activity by inhibitors induces simultaneous proteasomal degradation of many deregulated oncoproteins that are critical for all fundamental hallmarks of cancers. To date, mainly Hsp90 N-terminal domain inhibitors have been developed for cancer treatments; however, several of these have not been successful in clinical trials, often due to toxicity. One of the major drawbacks associated with Hsp90 N-terminal domain inhibition is induction of the pro-survival heat-shock response. This response results in increased levels of Hsp90 and anti-apoptotic proteins, such as heat shock factor 1 (HSF-1), Hsp27 and Hsp70, which initiate anti-apoptotic cascades and promote drug resistance, a highly problematic property of any anticancer drug. In contrast, Hsp90 C-terminal inhibitors do not induce the heat shock response, and are therefore promising novel agents for treatment of different cancers.
- Hsp90
- anticancer
- allosteric inhibitor
- virtual screening
- pharmacophore modelling
- molecular dynamics
1. Introduction
Cancer remains one of the leading causes of disease, mortality and economic loss worldwide [1]. While there have been considerable improvements in treatment and survival times for patients suffering from a number of different types of cancer, there is still an urgent need to identify novel molecular targets, curative therapies and anticancer agents with improved efficacy and reduced adverse outcomes [2][3]. Hsp90 is a highly conserved molecular chaperone that is responsible for the folding, activation and maturation of more than 300 client proteins, including protein kinases, E3-ligases and transcription factors [4][5]. Since these Hsp90 clients are involved and play important roles in cancer, Hsp90 has been recognized as a promising target for development of anticancer drugs [6]. Hsp90 is a homodimer composed of the N-terminal domain (NTD) with ATPase activity, the middle domain important for client and co-chaperone interactions, and the C-terminal domain (CTD), which mediates dimerization [5][7]. All Hsp90 inhibitors that have entered into clinical trials for the treatment of cancer bind to the ATP-binding site at Hsp90 NTD, and have displayed significant limitations, including unwanted side effects related to the induction of the heat shock response, as well as limited efficacy and dose-limiting toxicities, such as hepatotoxicity [8][9][10]. One of the major drawbacks associated with ATP-competitive Hsp90 NTD inhibition is the induction of the pro-survival heat shock response, which occurs at the same concentration that is required to outcompete the abundant ATP present in the cellular environment and to induce client degradation. The heat shock response results in increased levels of Hsp90 and anti-apoptotic proteins, such as heat shock factor 1 (HSF-1), Hsp27, and Hsp70, which initiate anti-apoptotic cascades and promote drug resistance [11]. Consequently, alternative mechanisms, such as selective Hsp90β inhibition [12], allosteric Hsp90 CTD inhibition [13], covalent Hsp90 inhibition [14], and/or targeting protein-protein interactions between Hsp90 and its co-chaperones [15], are promising new approaches being pursued [16].
The Hsp90 CTD contains a secondary nucleotide-binding site, which only becomes available after ATP binds to the Hsp90 NTD [17][18][19]. The aminocoumarin antibiotic novobiocin (Figure 1, compound 1) was the first compound shown to bind the Hsp90 CTD and induce the proteasomal degradation of oncogenic clients, such as Raf-1, v-Src, Her2 and mutated p53, by inhibiting their correct folding [17][20][21]. In contrast to Hsp90 NTD inhibitors, novobiocin did not induce the heat shock response after the treatment of cancer cells at concentrations needed for client protein degradation [17]. Since its discovery, several focused libraries have been synthesized to improve efficacy and explore the structure-activity relationships of CTD inhibitors. For example, replacement of the noviose sugar with ionizable amines (Figure 1, compounds 2 and 3), resulted in significant improvements in activity against various cancer cell lines [22][23]. In addition, the coumarin core of 2 could be replaced with biphenyl substituents (3) without losses of efficacy. Other natural products (e.g., epigallocatechin 3-gallate, coumermycin A1 and deguelin (Figure 1, compound 4)) also exhibited antiproliferative activity through Hsp90 CTD inhibition [13].
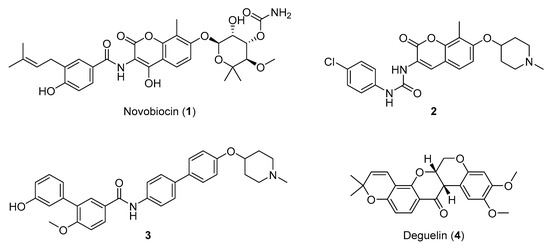
Figure 1. Representative Hsp90 C-terminal domain inhibitors. First Hsp90 C-terminal domain (CTD) inhibitor 1 (IC50 ~700 μM SKBr3 cancer cells) [17][21], synthetic novobiocin derivatives 2 and 3 (IC50 < 1 μM SKBr3 cancer cells) [22][23] and deguelin (4), a flavin natural product.
Though the efficacies of natural product analogues were much improved compared to the parent compounds, so far none have been co-crystallized with the Hsp90 CTD, limiting structure-based design approaches. In fact, whereas the Hsp90 NTD ATP-binding pocket is well described and several crystal structures with ATP and inhibitors have been published, the exact location of the allosteric pocket in Hsp90 CTD remains unknown. Several approaches to predict the Hsp90 CTD binding site using both experimental and molecular modelling methods have been reported.
2. Identification of the Hsp90 Allosteric Binding Site at C-Terminal Domain
The full-length Hsp90β cryoEM structure with bound ATP (PDB Code: 5FWK) [24] was used for CTD binding site prediction. The pocket finder algorithm in LigandScout 4.3 identified several binding pockets in the structure, as shown in Figure 2a. The orange isosurfaces represent pockets that were recommended by the program as druggable. The ATP binding sites were correctly identified at the Hsp90 NTD of each monomer, while a third large binding pocket was prioritized at the interface between two Hsp90 CTDs. The third allosteric pocket (Figure 2b) was selected for docking studies, MD simulations with novobiocin and compound 2, and the generation of SB pharmacophore models. It was delineated by residues 489, 603–613, 660–673 and 676 (monomer A), and residues 489–493, 515, 516, 601–609 and 663–677 (monomer B), and was similar to an allosteric binding site identified previously in yeast Hsp82 [25].
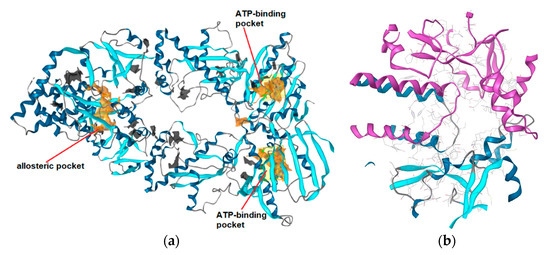
Figure 2. (a) Identification of putative binding pockets in Hsp90β cryoEM structure (PDB Code: 5FWK). The orange isosurfaces represent prioritized pockets identified by LigandScout 4.3. Two predicted pockets correspond to the ATP-binding sites in N-terminal domain of each Hsp90 monomer. A third large orange isosurface represents the allosteric pocket at the interface of two C-terminal domains. (b) Predicted allosteric pocket in the Hsp90 CTD at the interface of monomer A (magenta) and monomer B (blue) that was prioritized for docking, molecular dynamics and structure-based (SB)-pharmacophore modeling studies.
3. Binding Modes of Novobiocin and Compound 2 in the Hsp90 CTD Allosteric Pocket
The docked poses prioritized for the MD simulations of both compounds are shown in Figure 3.
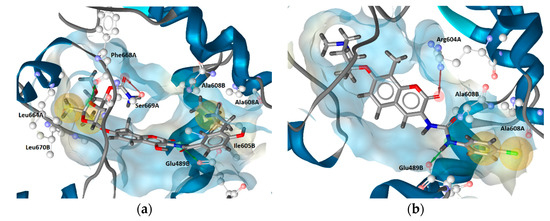
Figure 3. Prioritized docked poses of (a) novobiocin and (b) compound 2 in a predicted putative allosteric pocket in the Hsp90β C-terminal domain derived from the cryo-EM structure (PDB Code: 5FWK). For clarity, only amino acids interacting with respective compounds are shown. Hydrophobic features are shown as yellow spheres, H-bond donor as green arrow and H-bond acceptor as red arrow indicating a defined direction for H-bonding. The prioritized poses were used as starting points for molecular dynamics-based (MD) simulations.
A combined approach using molecular docking, molecular dynamics simulations and pharmacophore modelling was used to identify new Hsp90 CTD inhibitors (Figure 4). Virtual hits identified by virtual screening with both structure- and ligand-based pharmacophore models were tested for antiproliferative activity. Compound 11 (Figure 4), displayed antiproliferative activities in MCF-7 and Hep G2 cancer cell lines, inhibited Hsp90-dependent refolding of denatured luciferase and induced the degradation of Hsp90 clients without concomitant induction of Hsp70 levels.
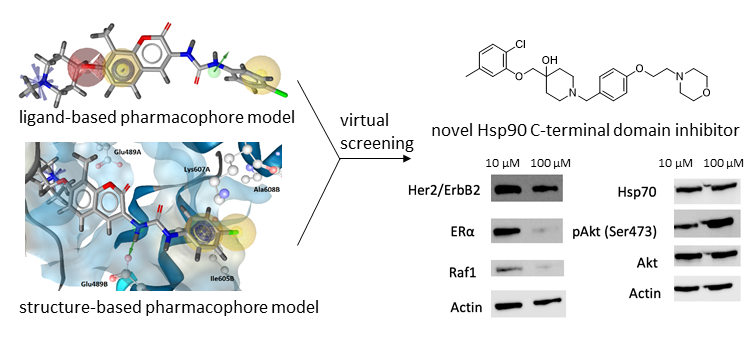
Figure 4. Discovery of novel structural class of allosteric Hsp90 CTD inhibitor using a combination of molecular modelling approaches.
References
- World Cancer Report—IARC. Available online: https://www.iarc.fr/cards_page/world-cancer-report/ (accessed on 19 August 2020).
- Dobbelstein, M.; Moll, U. Targeting tumour-supportive cellular machineries in anticancer drug development. Nat. Rev. Drug Discov. 2014, 13, 179–196.
- Baldo, P.; Fornasier, G.; Ciolfi, L.; Sartor, I.; Francescon, S. Pharmacovigilance in oncology. Int. J. Clin. Pharm. 2018, 40, 832–841.
- Prodromou, C.; Pearl, L.H. Structure and functional relationships of Hsp90. Curr. Cancer Drug Targets 2003, 3, 301–323.
- Röhl, A.; Rohrberg, J.; Buchner, J. The chaperone Hsp90: Changing partners for demanding clients. Trends Biochem. Sci. 2013, 38, 253–262.
- Trepel, J.; Mollapour, M.; Giaccone, G.; Neckers, L. Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer 2010, 10, 537–549.
- Pearl, L.H.; Prodromou, C. Structure and mechanism of the Hsp90 molecular chaperone machinery. Annu. Rev. Biochem. 2006, 75, 271–294.
- Yuno, A.; Lee, M.-J.; Lee, S.; Tomita, Y.; Rekhtman, D.; Moore, B.; Trepel, J.B. Clinical evaluation and biomarker profiling of Hsp90 inhibitors. Methods Mol. Biol. 2018, 1709, 423–441.
- Khandelwal, A.; Crowley, V.M.; Blagg, B.S.J. Natural product inspired N-terminal Hsp90 inhibitors: From bench to bedside? Med. Res. Rev. 2016, 36, 92–118.
- Neckers, L.; Workman, P. Hsp90 molecular chaperone inhibitors: Are we there yet? Clin. Cancer Res. 2012, 18, 64–76.
- Neckers, L.; Blagg, B.; Haystead, T.; Trepel, J.B.; Whitesell, L.; Picard, D. Methods to validate Hsp90 inhibitor specificity, to identify off-target effects, and to rethink approaches for further clinical development. Cell Stress Chaperones 2018, 23, 467–482.
- Khandelwal, A.; Kent, C.N.; Balch, M.; Peng, S.; Mishra, S.J.; Deng, J.; Day, V.W.; Liu, W.; Subramanian, C.; Cohen, M.; et al. Structure-guided design of an Hsp90β N-terminal isoform-selective inhibitor. Nat. Commun. 2018, 9, 1–7.
- Ferraro, M.; D’Annessa, I.; Moroni, E.; Morra, G.; Paladino, A.; Rinaldi, S.; Compostella, F.; Colombo, G. Allosteric modulators of HSP90 and HSP70: Dynamics meets function through structure-based drug design. J. Med. Chem 2019, 62, 60–87.
- Cuesta, A.; Wan, X.; Burlingame, A.L.; Taunton, J. Ligand conformational bias drives enantioselective modification of a surface-exposed lysine on Hsp90. J. Am. Chem. Soc. 2020, 142, 3392–3400.
- Wang, L.; Zhang, L.; Li, L.; Jiang, J.; Zheng, Z.; Shang, J.; Wang, C.; Chen, W.; Bao, Q.; Xu, X.; et al. Small-molecule inhibitor targeting the Hsp90-Cdc37 protein-protein interaction in colorectal cancer. Sci. Adv. 2019, 5, eaax2277.
- Li, L.; Wang, L.; You, Q.-D.; Xu, X.-L. Heat shock protein 90 inhibitors: An update on achievements, challenges, and future directions. J. Med. Chem. 2020, 63, 1798–1822.
- Marcu, M.G.; Chadli, A.; Bouhouche, I.; Catelli, M.; Neckers, L.M. The heat shock protein 90 antagonist novobiocin interacts with a previously unrecognized ATP-binding domain in the carboxyl terminus of the chaperone. J. Biol. Chem. 2000, 275, 37181–37186.
- Garnier, C.; Lafitte, D.; Tsvetkov, P.O.; Barbier, P.; Leclerc-Devin, J.; Millot, J.-M.; Briand, C.; Makarov, A.A.; Catelli, M.G.; Peyrot, V. Binding of ATP to heat shock protein 90: Evidence for an ATP-binding site in the C-terminal domain. J. Biol. Chem. 2002, 277, 12208–12214.
- Soti, C.; Vermes, A.; Haystead, T.A.J.; Csermely, P. Comparative analysis of the ATP-binding sites of Hsp90 by nucleotide affinity cleavage: A distinct nucleotide specificity of the C-terminal ATP-binding site. Eur. J. Biochem. 2003, 270, 2421–2428.
- Söti, C.; Rácz, A.; Csermely, P. A nucleotide-dependent molecular switch controls ATP binding at the C-terminal domain of Hsp90 N-terminal nucleotide binding unmasks a C-terminal binding pocket. J. Biol. Chem. 2002, 277, 7066–7075.
- Marcu, M.G.; Schulte, T.W.; Neckers, L. Novobiocin and related coumarins and depletion of heat shock protein 90-dependent signaling proteins. J. Natl. Cancer Inst. 2000, 92, 242–248.
- Garg, G.; Zhao, H.; Blagg, B.S.J. Design, synthesis and biological evaluation of alkylamino biphenylamides as Hsp90 C-terminal inhibitors. Bioorg. Med. Chem. 2017, 25, 451–457.
- Moroni, E.; Zhao, H.; Blagg, B.S.J.; Colombo, G. Exploiting conformational dynamics in drug discovery: Design of C-terminal inhibitors of Hsp90 with improved activities. J. Chem. Inf. Model. 2014, 54, 195–208.
- Matts, R.L.; Dixit, A.; Peterson, L.B.; Sun, L.; Voruganti, S.; Kalyanaraman, P.; Hartson, S.D.; Verkhivker, G.M.; Blagg, B.S.J. Elucidation of the Hsp90 C-terminal inhibitor binding site. ACS Chem. Biol. 2011, 6, 800–807.
- Morra, G.; Neves, M.A.C.; Plescia, C.J.; Tsustsumi, S.; Neckers, L.; Verkhivker, G.; Altieri, D.C.; Colombo, G. Dynamics-based discovery of allosteric inhibitors: Selection of new ligands for the C-terminal domain of Hsp90. J. Chem. Theory Comput. 2010, 6, 2978–2989.