Emerging evidence suggests that neuroinflammation is involved in both depression and neurodegenerative diseases. The kynurenine pathway, generating metabolites which may play a role in pathogenesis, is one of several competing pathways of tryptophan metabolism. A disturbed tryptophan metabolism with increased activity of the kynurenine pathway and production of quinolinic acid may result in deficiencies in tryptophan and derived neurotransmitters. Quinolinic acid is an N-methyl-D-aspartate receptor agonist, and raised levels in CSF, together with increased levels of inflammatory cytokines, have been reported in mood disorders. Increased quinolinic acid has also been observed in neurodegenerative diseases, including Parkinson’s disease, Alzheimer’s disease, amyotrophic lateral sclerosis, and HIV-related cognitive decline.
- tryptophan
- quinolinic acid
- neurodegeneration
- NMDA receptor
- depression
- Alzheimer
- Parkinson
- AIDS
- antioxidants
- glutathione
1. Introduction
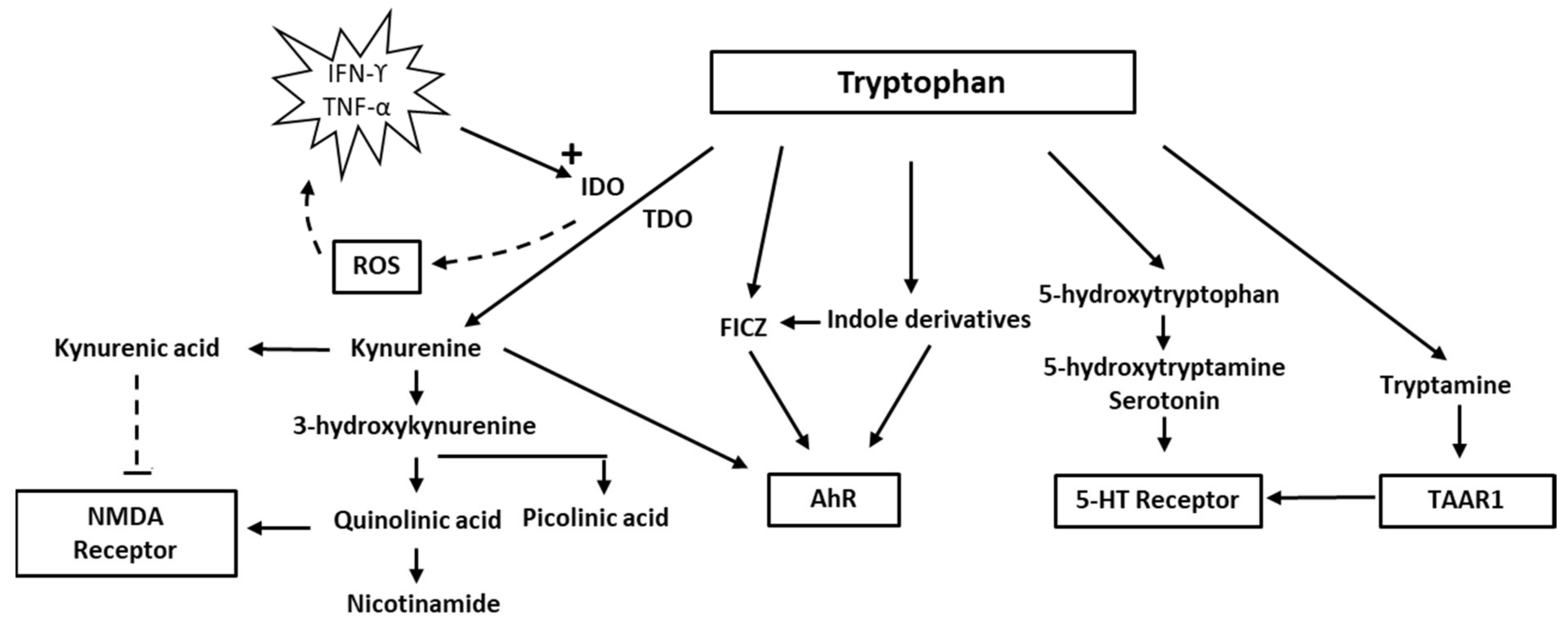
2. Tryptophan and Indolamine Dioxygenase
Indoleamine 2,3-dioxygenase belongs to the family of oxidoreductases, which also includes tryptophan 2,3-dioxygenase (TDO), sometimes referred to as tryptophan oxygenase. Both TDO and IDO contain a non-covalently bound Fe(II) heme unit per monomer. TDO is usually tetrameric, whereas IDO is monomeric. Several researchers [14,15][14][15] have discussed the mechanisms of tryptophan oxidation. It is assumed that it involves catalytic activity of the Fe(II) heme moiety of the enzymes and that TDO and IDO react by similar mechanisms. Because the interaction of tryptophan with oxygen appears to include the formation of an intermediary ferryl compound that may act as a strong oxidant [16], the IDO reaction may contribute to oxidative stress [17,18][17][18]. ROS may lead to increased production of inflammatory cytokines (TNF-alpha and IL-6, i.e., from macrophages), further upregulating IDO and thereby instigating a vicious circle [19]. The harmful effects of ROS formed from the IDO reaction may be counteracted by such endogenous antioxidants as the glutathione (GSH)/GSH-peroxidase system and presumably also by such exogenous antioxidants as polyphenols. Increased production of peroxide and ROS resulting from IDO activation may conceivably disrupt the finely tuned interaction between intracellular oxidants and antioxidants, concurrent with a change in the tryptophan/kynurenine ratio. Overexpression of IDO, leading to an accelerated tryptophan–kynurenine pathway and ROS production, may cause a depressed intracellular concentration of reduced GSH and disrupt the intracellular redox balance [20]. Upon interaction with ROS, intracellular GSH is rapidly oxidised to its disulfide form GSSG [21], which can either be reduced again enzymatically to GSH intracellularly or expelled from the cells [22[22][23],23], where it may form S-glutathionylated plasma proteins [24] or lead to increased extracellular levels of its amino acid constituents [25], which can be detected in blood [26]. Under the redox conditions existing in circulating blood, not only oxidised GSH, but also cysteine and homocysteine, will exist predominantly as mixed disulfides with albumin or other plasma proteins [27,28][27][28]. Kynurenine and its breakdown products, such as quinolinic acid, have diverse biological impacts, including immunological and psychological effects [11,29,30][11][29][30]. In addition, the interferon (IFN-α) treatment for hepatitis C may precipitate depressive symptoms, due to activation of the kynurenine pathway [31,32][31][32]. Of relevance also is the fact that kynurenine by the enzyme kynurenine-3-hydroxylase can be converted to 3-hydroxy-kynurenine, which, in addition to being a redox reactive metabolite, appears to act as a neurotoxin through its ability to induce protein modifications [33]. In addition, activation of the kynurenine pathway appears to enhance the development of cardiovascular disease [34,35][34][35]. Until recent years, medical interest in tryptophan has focused mainly on its role in serotonin synthesis. In the CNS, serotonin modulates mood and cognition. Several neuropsychiatric problems have been attributed to changes in the availability of serotonin. The focus has currently changed, however, to other aspects of tryptophan metabolism such as the indole and kynurenine pathways. There is a delicate balance between different tryptophan pathways. It is now realised that only about 1–2% of tryptophan intake is metabolised into serotonin and approximately 95% is metabolised via the kynurenine pathway. Because the two metabolic pathways of serotonin and kynurenine compete for the substrate, tryptophan, increased metabolism of tryptophan via the kynurenine pathway may result in a relative deficiency of serotonin in the CNS. Thus, people vulnerable to stress may benefit from tryptophan supplements [36]. Notably, immune activation accompanied by increased production of INFγ will increase both TDO and IDO activity and thereby channel the tryptophan metabolism into the kynurenine pathway. IDO oxidation stimulated by immune activation is apparently of significance for a variety of disorders, including neuropsychological impairments and diseases with vascular pathology [11,35][11][35].3. Quinolinic Acid
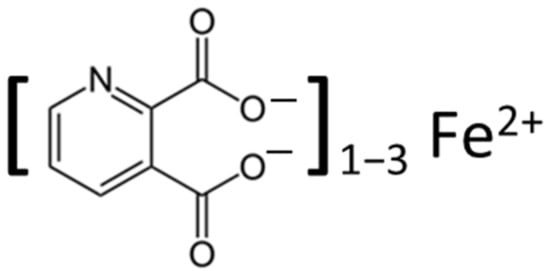
Quinolinic acid [37], also known as pyridine-2,3-dicarboxylic acid, is a dicarboxylic acid with a pyridine backbone—a downstream product of the kynurenine pathway (Figure 1 and Figure 2). It is known that quinolinic acid can act as an N-methyl-d-aspartate (NMDA) receptor agonist [38,39][38][39].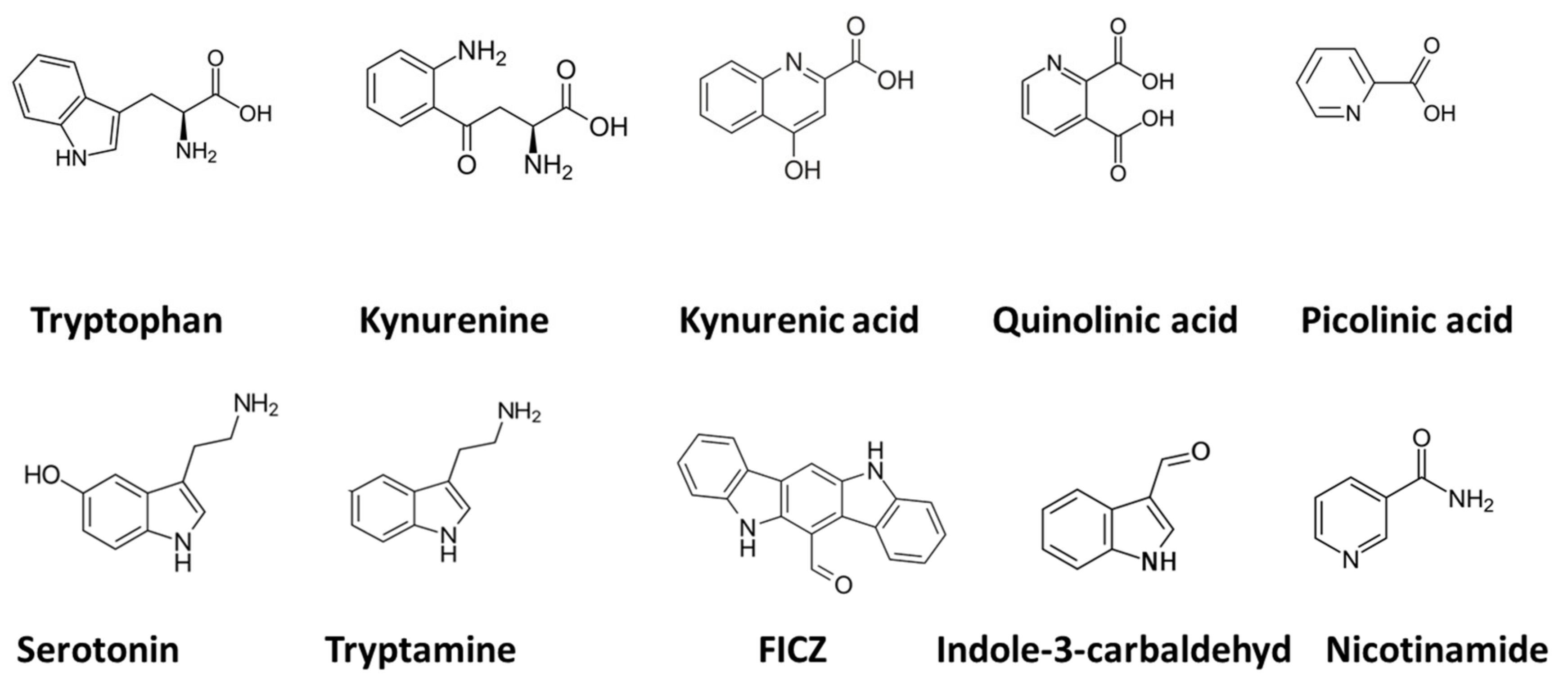
3.1. Mood Disorders: Depression
In post-mortem brains of patients who had suffered from major depression, the levels of quinolinic acid in the prefrontal cortex have been found to be greater than in post-mortem brains of patients without depression [53]. Because NMDA receptor antagonists possess antidepressant properties, it is tempting to propose that increased levels of quinolinic acid in patients with depression may serve a causal or aggravating role in the mood disorder through the activation of NMDA receptors. Following INFα therapy, researchers have found increased concentrations of quinolinic acid in the cerebrospinal fluid (CSF) and have noted that the concentrations correlate with the severity of depressive symptoms [53]. In addition, increased levels of quinolinic acid could play a role in impairment of the glial–neuronal network, which could be associated with the recurrent and chronic nature of bipolar depression [54]. Based on results of a meta-analysis, Marx and co-workers (2021) have suggested that there is a shift in the tryptophan metabolism from serotonin to the kynurenine pathway in psychiatric diseases and that patients with mood disorders are characterised by a higher quinolinic-acid/kynurenic-acid ratio than people without mood disorders [55]. Moreover, Brundin and co-workers (2016) found a higher quinolinic-acid/picolinic-acid ratio in the CSF of 137 patients who were exhibiting suicidal behaviour, as compared with the values in 71 healthy controls [56]. They ascribed their observation to a reduction of an alternative kynurenine pathway metabolite, picolinic acid, that counteracts the neurotoxic action of quinolinic acid [57]. The reduced concentrations of the protective metabolite, picolinic acid, in CSF were sustained over two years after a suicide attempt in depressive patients. Although kynurenic acid, another metabolite of kynurenine, was not assessed in the latter study [57], it might have worked to counteract excessive NMDA receptor activation and exert a neuroprotective action [58,59,60,61][58][59][60][61]. An antidepressant effect of kynurenic acid is in accordance with the results from an experimental study in a rat model [62].References
- Kanova, M.; Kohout, P. Tryptophan: A Unique Role in the Critically Ill. Int. J. Mol. Sci. 2021, 22, 11714.
- Melhem, N.J.; Taleb, S. Tryptophan: From Diet to Cardiovascular Diseases. Int. J. Mol. Sci. 2021, 22, 9904.
- Gainetdinov, R.R.; Hoener, M.C.; Berry, M.D. Trace Amines and Their Receptors. Pharmacol. Rev. 2018, 70, 549–620.
- Roth, W.; Zadeh, K.; Vekariya, R.; Ge, Y.; Mohamadzadeh, M. Tryptophan Metabolism and Gut-Brain Homeostasis. Int. J. Mol. Sci. 2021, 22, 2973.
- Tanaka, M.; Spekker, E.; Szabó, Á.; Polyák, H.; Vécsei, L. Modelling the neurodevelopmental pathogenesis in neuropsychiatric disorders. Bioactive kynurenines and their analogues as neuroprotective agents-in celebration of 80th birthday of Professor Peter Riederer. J. Neural Transm. 2022, 129, 627–642.
- Ramírez Ortega, D.; Ugalde Muñiz, P.E.; Blanco Ayala, T.; Vázquez Cervantes, G.I.; Lugo Huitrón, R.; Pineda, B.; González Esquivel, D.F.; Pérez de la Cruz, G.; Pedraza Chaverrí, J.; Sánchez Chapul, L.; et al. On the Antioxidant Properties of L-Kynurenine: An Efficient ROS Scavenger and Enhancer of Rat Brain Antioxidant Defense. Antioxidants 2021, 11, 31.
- Werner, E.R.; Werner-Felmayer, G. Substrate and cofactor requirements of indoleamine 2,3-dioxygenase in interferon-gamma-treated cells: Utilization of oxygen rather than superoxide. Curr. Drug Metab. 2007, 8, 201–203.
- Blunt, C.E.; Torcuk, C.; Liu, Y.; Lewis, W.; Siegel, D.; Ross, D.; Moody, C.J. Synthesis and Intracellular Redox Cycling of Natural Quinones and Their Analogues and Identification of Ind.doleamine-2,3-dioxygenase (IDO) as Potential Target for Anticancer Activity. Angew. Chem. Int. Ed. Engl. 2015, 54, 8740–8745.
- Tauil, C.B.; da Rocha Lima, A.D.; Ferrari, B.B.; da Silva, V.A.G.; Moraes, A.S.; da Silva, F.M.; Melo-Silva, C.A.; Farias, A.S.; Brandão, C.O.; Leonilda, M.B.D.; et al. Depression and anxiety in patients with multiple sclerosis treated with interferon-beta or fingolimod: Role of indoleamine 2,3-dioxygenase and pro-inflammatory cytokines. Brain Behav. Immun. Health 2020, 9, 100162.
- Hestad, K.A.; Aukrust, P.; Tønseth, S.; Reitan, S.K. Depression has a Strong Relationship to Alterations in the Immune, Endocrine and Neural System. Curr. Psychiatry Rev. 2009, 5, 287–297.
- Hestad, K.A.; Engedal, K.; Whist, J.E.; Farup, P.G. The Relationships among Tryptophan, Kynurenine, Indoleamine 2,3-Dioxygenase, Depression, and Neuropsychological Performance. Front. Psychol. 2017, 8, 1561.
- Mangge, H.; Summers, K.L.; Meinitzer, A.; Zelzer, S.; Almer, G.; Prassl, R.; Schnedl, W.J.; Reininghaus, E.; Paulmichl, K.; Weghuber, D.; et al. Obesity-related dysregulation of the tryptophan-kynurenine metabolism: Role of age and parameters of the metabolic syndrome. Obesity 2014, 22, 195–201.
- Favennec, M.; Hennart, B.; Caiazzo, R.; Leloire, A.; Yengo, L.; Verbanck, M.; Arredouani, A.; Marre, M.; Pigeyre, M.; Bessede, A.; et al. The kynurenine pathway is activated in human obesity and shifted toward kynurenine monooxygenase activation. Obesity 2015, 23, 2066–2074.
- Lewis-Ballester, A.; Pham, K.N.; Batabyal, D.; Karkashon, S.; Bonanno, J.B.; Poulos, T.L.; Yeh, S.R. Structural insights into substrate and inhibitor binding sites in human indoleamine 2,3-dioxygenase 1. Nat. Commun. 2017, 8, 1693.
- Raven, E.L. A short history of heme dioxygenases: Rise, fall and rise again. J. Biol. Inorg. Chem. 2017, 22, 175–183.
- Lewis-Ballester, A.; Batabyal, D.; Egawa, T.; Lu, C.; Lin, Y.; Marti, M.A.; Capece, L.; Estrin, D.A.; Yeh, S.R. Evidence for a ferryl intermediate in a heme-based dioxygenase. Proc. Natl. Acad. Sci. USA 2009, 106, 17371–17376.
- Le Floc’h, N.; Otten, W.; Merlot, E. Tryptophan metabolism, from nutrition to potential therapeutic applications. Amino Acids 2011, 41, 1195–1205.
- Wichers, M.C.; Maes, M. The role of indoleamine 2,3-dioxygenase (IDO) in the pathophysiology of interferon-alpha-induced depression. J. Psychiatry Neurosci. 2004, 29, 11–17.
- Illán-Gómez, F.; Gonzálvez-Ortega, M.; Orea-Soler, I.; Alcaraz-Tafalla, M.S.; Aragón-Alonso, A.; Pascual-Díaz, M.; Pérez-Paredes, M.; Lozano-Almela, M.L. Obesity and inflammation: Change in adiponectin, C-reactive protein, tumour necrosis factor-alpha and interleukin-6 after bariatric surgery. Obes. Surg. 2012, 22, 950–955.
- Wang, Q.; Liu, D.; Song, P.; Zou, M.H. Tryptophan-kynurenine pathway is dysregulated in inflammation, and immune activation. Front. Biosci. 2015, 20, 1116–1143.
- Meister, A.; Anderson, M.E. Glutathione. Annu. Rev. Biochem. 1983, 52, 711–760.
- Srivastava, S.K.; Beutler, E. The transport of oxidized glutathione from human erythrocytes. J. Biol. Chem. 1969, 244, 9–16.
- Akerboom, T.P.; Bilzer, M.; Sies, H. The relationship of biliary glutathione disulfide efflux and intracellular glutathione disulfide content in perfused rat liver. J. Biol. Chem. 1982, 257, 4248–4252.
- Giustarini, D.; Milzani, A.; Dalle-Donne, I.; Rossi, R. Measurement of S-glutathionylated proteins by HPLC. Amino Acids 2021, 54, 675–686.
- Baudouin-Cornu, P.; Lagniel, G.; Kumar, C.; Huang, M.E.; Labarre, J. Glutathione degradation is a key determinant of glutathione homeostasis. J. Biol. Chem. 2012, 287, 4552–4561.
- Elshorbagy, A.K.; Nurk, E.; Gjesdal, C.G.; Tell, G.S.; Ueland, P.M.; Nygård, O.; Tverdal, A.; Vollset, S.E.; Refsum, H. Homocysteine, cysteine, and body composition in the Hordaland Homocysteine Study: Does cysteine link amino acid and lipid metabolism? Am. J. Clin. Nutr. 2008, 88, 738–746.
- Mansoor, M.A.; Svardal, A.M.; Ueland, P.M. Determination of the in vivo redox status of cysteine, cysteinylglycine, homocysteine, and glutathione in human plasma. Anal. Biochem. 1992, 200, 218–229.
- Giles, G.I.; Tasker, K.M.; Jacob, C. Hypothesis: The role of reactive sulfur species in oxidative stress. Free Radic. Biol. Med. 2001, 31, 1279–1283.
- Rajan, D.; Chinnadurai, R.; O’Keefe, E.L.; Boyoglu-Barnum, S.; Todd, S.O.; Hartert, T.V.; Galipeau, J.; Anderson, L.J. Protective role of Indoleamine 2,3 dioxygenase in Respiratory Syncytial Virus associated immune response in airway epithelial cells. Virology 2017, 512, 144–150.
- Tanaka, M.; Vécsei, L. Monitoring the kynurenine system: Concentrations, ratios or what else? Adv. Clin. Exp. Med. 2021, 30, 775–778.
- Oxenkrug, G.F. Interferon-gamma-inducible kynurenines/pteridines inflammation cascade: Implications for aging and aging-associated psychiatric and medical disorders. J. Neural Transm. 2011, 118, 75–85.
- Hunt, C.; Macedo, E.C.T.; Suchting, R.; de Dios, C.; Cuellar Leal, V.A.; Soares, J.C.; Dantzer, R.; Teixeira, A.L.; Selvaraj, S. Effect of immune activation on the kynurenine pathway and depression symptoms—A systematic review and meta-analysis. Neurosci. Biobehav. Rev. 2020, 118, 514–523.
- Capucciati, A.; Galliano, M.; Bubacco, L.; Zecca, L.; Casella, L.; Monzani, E.; Nicolis, S. Neuronal Proteins as Targets of 3-Hydroxykynurenine: Implications in Neurodegenerative Diseases. ACS Chem. Neurosci. 2019, 10, 3731–3739.
- Sulo, G.; Vollset, S.E.; Nygård, O.; Midttun, Ø.; Ueland, P.M.; Eussen, S.J.; Pedersen, E.R.; Tell, G.S. Neopterin and kynurenine-tryptophan ratio as predictors of coronary events in older adults, the Hordaland Health Study. Int. J. Cardiol. 2013, 168, 1435–1440.
- Zuo, H.; Ueland, P.M.; Ulvik, A.; Eussen, S.J.; Vollset, S.E.; Nygård, O.; Midttun, Ø.; Theofylaktopoulou, D.; Meyer, K.; Tell, G.S. Plasma Biomarkers of Inflammation, the Kynurenine Pathway, and Risks of All-Cause, Cancer, and Cardiovascular Disease Mortality: The Hordaland Health Study. Am. J. Epidemiol. 2016, 183, 249–258.
- Markus, C.R.; Verschoor, E.; Firk, C.; Kloek, J.; Gerhardt, C.C. Effect of tryptophan-rich egg protein hydrolysate on brain tryptophan availability, stress and performance. Clin. Nutr. 2010, 29, 610–616.
- Carrillo-Mora, P.; Pérez-De la Cruz, V.; Estrada-Cortés, B.; Toussaint-González, P.; Martínez-Cortéz, J.A.; Rodríguez-Barragán, M.; Quinzaños-Fresnedo, J.; Rangel-Caballero, F.; Gamboa-Coria, G.; Sánchez-Vázquez, I.; et al. Serum Kynurenines Correlate with Depressive Symptoms and Disability in Poststroke Patients: A Cross-sectional Study. Neurorehabil. Neural Repair. 2020, 34, 936–944.
- Misztal, M.; Frankiewicz, T.; Parsons, C.G.; Danysz, W. Learning deficits induced by chronic intraventricular infusion of quinolinic acid--protection by MK-801 and memantine. Eur. J. Pharmacol. 1996, 296, 1–8.
- St’astný, F.; Lisý, V.; Mares, V.; Lisá, V.; Balcar, V.J.; Santamaría, A. Quinolinic acid induces NMDA receptor-mediated lipid peroxidation in rat brain microvessels. Redox Rep. 2004, 9, 229–233.
- Wu, W.; Nicolazzo, J.A.; Wen, L.; Chung, R.; Stankovic, R.; Bao, S.S.; Lim, C.K.; Brew, B.J.; Cullen, K.M.; Guillemin, G.J. Expression of tryptophan 2,3-dioxygenase and production of kynurenine pathway metabolites in triple transgenic mice and human Alzheimer’s disease brain. PLoS ONE 2013, 8, e59749.
- Lugo-Huitrón, R.; Ugalde Muñiz, P.; Pineda, B.; Pedraza-Chaverrí, J.; Ríos, C.; Pérez-de la Cruz, V. Quinolinic acid: An endogenous neurotoxin with multiple targets. Oxid. Med. Cell. Longev. 2013, 2013, 104024.
- Guillemin, G.J.; Smith, D.G.; Smythe, G.A.; Armati, P.J.; Brew, G.J. Expression of the kynurenine pathway enzymes in human microglia and macrophages. Adv. Exp. Med. Biol. 2003, 527, 105–112.
- Guillemin, G.J.; Cullen, K.M.; Lim, C.K.; Smythe, G.A.; Garner, B.; Kapoor, V.; Takikawa, O.; Brew, B.J. Characterization of the kynurenine pathway in human neurons. J. Neurosci. 2007, 27, 12884–12892.
- Heyes, M.P.; Achim, C.L.; Wiley, C.A.; Major, E.O.; Saito, K.; Markey, S.P. Human microglia convert l-tryptophan into the neurotoxin quinolinic acid. Biochem. J. 1996, 320 Pt 2, 595–597.
- Heyes, M.P.; Saito, K.; Lackner, A.; Wiley, C.A.; Achim, C.L.; Markey, S.P. Sources of the neurotoxin quinolinic acid in the brain of HIV-1-infected patients and retrovirus-infected macaques. FASEB J. 1998, 12, 881–896.
- Schwarcz, R.; Bruno, J.P.; Muchowski, P.J.; Wu, H.Q. Kynurenines in the mammalian brain: When physiology meets pathology. Nat. Rev. Neurosci. 2012, 13, 465–477.
- La Cruz, V.P.D.; Carrillo-Mora, P.; Santamaría, A. Quinolinic Acid, an endogenous molecule combining excitotoxicity, oxidative stress and other toxic mechanisms. Int. J. Tryptophan Res. 2012, 5, 1–8.
- Verma, M.K.; Goel, R.; Nandakumar, K.; Nemmani, K.V. Bilateral quinolinic acid-induced lipid peroxidation, decreased striatal monoamine levels and neurobehavioral deficits are ameliorated by GIP receptor agonist D-Ala(2)GIP in rat model of Huntington’s disease. Eur. J. Pharmacol. 2018, 828, 31–41.
- Pierozan, P.; Pessoa-Pureur, R. Cytoskeleton as a Target of Quinolinic Acid Neurotoxicity: Insight from Animal Models. Mol. Neurobiol. 2018, 55, 4362–4372.
- Zhou, Q.; Sheng, M. NMDA receptors in nervous system diseases. Neuropharmacology 2013, 74, 69–75.
- Kubicova, L.; Hadacek, F.; Weckwerth, W.; Chobot, V. Effects of endogenous neurotoxin quinolinic acid on reactive oxygen species production by Fenton reaction catalyzed by iron or copper. J. Organomet. Chem. 2015, 782, 111–115.
- Aaseth, J.; Crisponi, G.; Anderson, O. Chelation Therapy in the Treatment of Metal Intoxication; Academic Press: Cambridge, MA, USA, 2016.
- Myint, A.M. Kynurenines: From the perspective of major psychiatric disorders. FEBS J. 2012, 279, 1375–1385.
- Maes, M.; Leonard, B.E.; Myint, A.M.; Kubera, M.; Verkerk, R. The new ‘5-HT’ hypothesis of depression: Cell-mediated immune activation induces indoleamine 2,3-dioxygenase, which leads to lower plasma tryptophan and an increased synthesis of detrimental tryptophan catabolites (TRYCATs), both of which contribute to the onset of depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 2011, 35, 702–721.
- Marx, W.; McGuinness, A.J.; Rocks, T.; Ruusunen, A.; Cleminson, J.; Walker, A.J.; Gomes-da-Costa, S.; Lane, M.; Sanches, M.; Diaz, A.P.; et al. The kynurenine pathway in major depressive disorder, bipolar disorder, and schizophrenia: A meta-analysis of 101 studies. Mol. Psychiatry 2021, 26, 4158–4178.
- Brundin, L.; Sellgren, C.M.; Lim, C.K.; Grit, J.; Pålsson, E.; Landén, M.; Samuelsson, M.; Lundgren, K.; Brundin, P.; Fuchs, D.; et al. An enzyme in the kynurenine pathway that governs vulnerability to suicidal behavior by regulating excitotoxicity and neuroinflammation. Transl. Psychiatry 2016, 6, e865.
- Beninger, R.J.; Colton, A.M.; Ingles, J.L.; Jhamandas, K.; Boegman, R.J. Picolinic acid blocks the neurotoxic but not the neuroexcitant properties of quinolinic acid in the rat brain: Evidence from turning behaviour and tyrosine hydroxylase immunohistochemistry. Neuroscience 1994, 61, 603–612.
- Guillemin, G.J.; Smythe, G.; Takikawa, O.; Brew, B.J. Expression of indoleamine 2,3-dioxygenase and production of quinolinic acid by human microglia, astrocytes, and neurons. Glia 2005, 49, 15–23.
- Chen, Y.; Guillemin, G.J. Kynurenine pathway metabolites in humans: Disease and healthy States. Int. J. Tryptophan Res. 2009, 2, 1–19.
- Martos, D.; Tuka, B.; Tanaka, M.; Vécsei, L.; Telegdy, G. Memory Enhancement with Kynurenic Acid and Its Mechanisms in Neurotransmission. Biomedicines 2022, 10, 849.
- Erabi, H.; Okada, G.; Shibasaki, C.; Setoyama, D.; Kang, D.; Takamura, M.; Yoshino, A.; Fuchikami, M.; Kurata, A.; Kato, T.A.; et al. Kynurenic acid is a potential overlapped biomarker between diagnosis and treatment response for depression from metabolome analysis. Sci. Rep. 2020, 10, 16822.
- Tanaka, M.; Bohár, Z.; Martos, D.; Telegdy, G.; Vécsei, L. Antidepressant-like effects of kynurenic acid in a modified forced swim test. Pharmacol. Rep. 2020, 72, 449–455.