DUX4-rearrangement (DUX4r) is a recently discovered recurrent genomic lesion reported in 4-7% of B cell acute lymphoblastic leukaemia (B-ALL). The gene fusion most commonly links the hypervariable IGH gene to DUX4 a gene located within the D4Z4 macrosatellite repeat on chromosome 4. DUX4r is cryptic to most standard diagnostic techniques and difficult to identify even with next-generation sequencing assays.
- acute lymphoblastic leukaemia
- DUX4
- ERG
- DUX4-IGH
- DUX4-rearragnement
Note:All the information in this draft can be edited by authors. And the entry will be online only after authors edit and submit it.
1. Introduction
B-cell acute lymphoblastic leukaemia (B-ALL) is a malignant disorder of the bone marrow resulting in over proliferation of immature B lymphoblasts. The disease can manifest at any age but the majority of patients are children, making B-ALL the most common childhood malignancy [1]. This heterogeneous disease is characterised by a variety of different genomic alterations including changes in chromosome number, chromosomal translocations and single nucleotide variants (SNV). Detection of the underlying genomic alterations assists clinical risk stratification and therapeutic triage. Cytogenetic analysis has proven adept at identifying several recurrent genomic alterations which result in diseases with distinct gene expression profiles (GEP) and defined prognosis. This includes high hyperdiploidy, hypodiploidy, and the translocations t(12;21) [ETV6-RUNX1], t(9;22) BCR-ABL1, t(1;19) TCF3-PBX1 and alterations of chromosome 11q23 resulting in rearrangement of KMT2A/MLL. These alterations account for approximately 60% of pediatric B-ALL cases [2,3][2][3]. Remaining patients were historically classified as B-other and demonstrated highly variable prognosis and treatment response.
Molecular studies involving GEP and next-generation sequencing (NGS) have subsequently identified a number of additional recurrent molecular alterations not detectable with standard cytogenetics, several of which may be targetable by precision medicine approaches. This includes the newly recognized subtype of Philadelphia chromosome-like (Ph-like) ALL characterized by a gene expression profile similar to cases with a BCR-ABL1 translocation, but instead carrying one of multiple kinase activating lesions. One example is rearrangement of the cytokine receptor gene CRLF2 (CRLF2r), commonly with concurrence of Janus Kinase 2 (JAK2) mutations [4,5][4][5], affecting 5–7% of children with B-ALL [3]. NGS has also recently identified a rearrangement of the homeodomain encoding the Double Homeobox 4 (DUX4) transcription factor with the immunoglobulin heavy chain (IGH) locus which results in a distinct genetic subtype.
As early as 2002 researchers identified a novel B-ALL subtype, with a distinct microarray GEP, not associated with any known recurrent genomic alterations, that appeared to confer a good prognosis. Interrogation of overexpressed genes in these patients failed to uncover a causative lesion [6]. Follow-up studies involving copy number alteration (CNA) analysis revealed many of the patients with the distinct expression profile also demonstrated deletion of the ERG gene (ETS transcription factor ERG), a genomic alteration absent in almost all other subtypes. ERG deletion was consequently proposed as the driving lesion in this subtype [7,8][7][8]. Multiple studies have subsequently demonstrated that monoallelic deletion of ERG is observed in only a subset of patients demonstrating this GEP. Furthermore, ERG deletions were subclonal in several patients at diagnosis and either altered or absent at relapse [9–11][9][10][11].
In 2016, two independent studies identified rearrangement of the DUX4 locus (DUX4r), most commonly partnered with IGH, present in patients with the previously detected GEP [12,13][12][13]. Transduction of the DUX4 fusion transcript into NIH3T3 fibroblasts resulted in cellular transformations, demonstrating the oncogenic potential of this alteration [12]. Multiple studies have subsequently confirmed the unique GEP of DUX4r cases and identified the rearrangement in 4–7% of B-ALL patients [9,14–19][9][14][15][16][17][18][19], as well as within the NALM6 cell line [12]. Leukaemic cells carrying the DUX4r also display a unique methylation profile, associated with widespread hypomethylation [15,20,21][15][20][21], and express a specific non coding RNA signature [16,21][16][21]. Consequently, DUX4r, also reported in the literature as DUX4/ERG, has increasingly been accepted as a distinct molecular subtype in B-ALL. In this review, we present the current understanding of the molecular structure and biological effects of the DUX4r.
2. Description of the DUX4 Rearrangement
The DUX4 gene is present within each repeat of the D4Z4 tandem array located in the subtelomeric region of chromosome 4q [22], with an almost identical locus (>98% nucleotide identity) on 10q [23,24][23][24]. The D4Z4 array is polymorphic in length containing between 11–100 copies of the 3.3 kb repeat in healthy adults (Figure 1) [25]. In healthy tissue, transcription of DUX4 is restricted to germline cells of the testes. Transcription has also been observed in induced pluripotent stem cells, suggesting a role for DUX4 in germline development [26,27][26][27]. Expression of the full-length DUX4 transcript is epigenetically silenced in somatic tissue [27]. Only the first exon of the spliced transcript contains a coding sequence for the protein which consists of two N-terminal homeodomains capable of DNA binding [28] and a C-terminal transactivation domain [29]. The DUX4 protein is capable of binding to, and upregulating expression of, multiple genes as well as initiating expression from alternate promoters, producing non-canonical transcript isoforms [29].
Contraction of the D4Z4 region resulting in fewer than 10 repeats is associated with facioscapulohumeral muscular dystrophy (FSHD) [25,30][25][30], a genetically inherited disorder that initially manifests as progressive weakening of the facial, shoulder and upper arm muscles [31]. Partial deletion of the D4Z4 array is associated with hypomethylation and loss of repressive histone modifications that are believed to reduce chromatin packing of the subtelomeric region allowing DUX4 expression [32,33][32][33]. Intriguingly, FSHD only manifests in patients who demonstrate D4Z4 contraction on chromosome 4q and not the homologous array on chromosome 10q. Furthermore, contraction of an alternative chromosome 4q allele (4qB) [24], does not result in disease. Sequencing efforts have subsequently revealed that the permissive 4qA allele carries a polymorphism in the region immediately distal to the final repeat of the array (Figure 1). This polymorphism creates a canonical polyadenylation signal in the 3’UTR of DUX4 enabling expression of a stable mRNA transcript [34]. The translated protein then binds multiple target genes resulting in widespread changes in gene expression that are ultimately cytotoxic [26].
DUX4 has also been implicated in several cancers involving rearrangements that produce chimeric proteins with altered transcriptional activity [35,36][35][36]. For example, a recurrent translocation between Capicua Transcriptional Repressor (CIC) and DUX4 occurs in a proportion of patients with Ewing-like sarcoma. This chromosomal rearrangement produces an in-frame transcript containing the first 20 exons of CIC but replacing the terminal exons with the 3’ portion of DUX4. Translation of the chimeric transcript produces a protein that retains the majority of CIC, including the N-terminal DNA binding domains, but replaces the C-terminus with the DUX4 transactivation domain. As a result, the chimeric CIC-DUX4 protein acts as an oncogenic transcriptional activator [37]. In B-ALL, translocation of DUX4 results in a different chimeric protein, but one that again acts as an oncogenic transcriptional activator. In all but one reported case, the 5’ coding sequence of DUX4 is cryptically inserted into an alternate genomic location, resulting in expression of a chimeric transcript which retains sequence containing the N-terminus of DUX4 but replacing the 3’ coding sequence (Figure 2). While multiple potential fusion partners have been identified, including ERG, DUX4 is most commonly inserted into the IGH locus [9,12,13,17][9][12][13][17].
Multiple rearrangements of IGH (IGHr) have been reported in B-ALL resulting in expression or overexpression of genes with oncogenic potential. The most common of these is a translocation between chromosome 14 and CRLF2 of the pseudoautosomal region of chromosome X/Y resulting in increased expression of cytokine receptor-like factor 2 [4,38][4][38]. In the case of DUX4, most analyses report that the rearrangement involves a portion of the D4Z4 array on 4q or the homologous region on 10q, consisting of either a partial copy of DUX4 or one complete and one partial D4Z4 repeat, being inserted into the IGH locus, placing them close to the IGH enhancer (E) [12]. As with other IGHr, the presence of the enhancer induces expression of the translocated gene [38]. Repeats containing DUX4 can be inserted in either orientation resulting in expression from the positive or negative strand. In some cases, a more complex rearrangement involving sequences from a third genomic location have been reported (Figure 1C) [12,15][12][15]. Alternatively, Hi-C data performed on the NALM6 cell line suggest that a reciprocal translocation can occur in which the telomeric ends of 4/10q are exchanged with 14q [39].
IGH breakpoints are enriched in the 3.5 kb region preceding the IGHM constant allele and overlapping the IGH D-J junctions but can occur throughout the locus [9]. Breakpoint locations for the DUX4 gene are harder to define given the repetitive nature of the D4Z4 array, but most commonly occur within the 5’ region upstream of DUX4 and within the 3’ coding region of exon1. This results in a DUX4 transcript which maintains the homeodomains encoded at the 5’ end of the transcript fused to sequence, usually from IGH-JH or IGH-DH regions, but can also be another genomic location [9,12,13,40][9][12][13][40]. The resulting protein thus maintains its ability to bind DUX4 targets but possesses a truncated C-terminus with inclusions of some amino acids encoded by the alternate locus. As the genomic breakpoints for this rearrangement are highly variable, the resultant length and amino acid sequence the C-terminal domain of the DUX4-fusion varies considerably between patients but consistently retains the DNA binding homeodomains (Figure 2) [9,12,39][9][12][39].
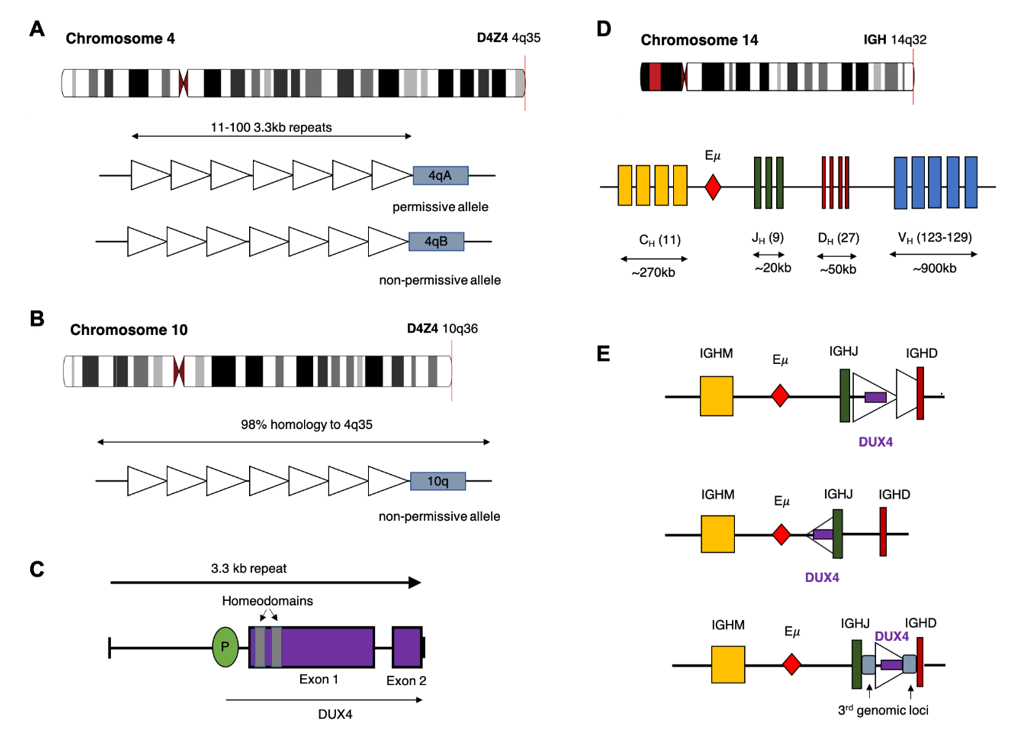
Figure 1. Potential chromosomal rearrangements involving IGH and DUX4. (A) Ideogram of chromosome 4 indicating location of the D4Z4 array and a depiction of the two alleles which vary in the sequence distal to the final repeat (repeat indicated by open triangles). This includes the permissive 4qA allele which can result in FSHD when contracted to fewer than 10 repeats. (B) Ideogram of chromosome 10 indicating location of the homologous D4Z4 array with 98% identical sequence. This chromosome is associated with a non-permissive allele which does not result in FSHD. (C) Schematic diagram of the repeat 3.3 kb repeat sequence indicating location and exons of the DUX4 gene. (D) Ideogram of chromosome 14 and depiction of the IGH locus indicating constant (CH), joining (JH), diversity (DH) and variable (VH) alleles (E) Schematic diagram depicting possible rearrangements as a result of cryptic insertion of DUX4 from either chromosome 4 or 10 into the IGH locus. DUX4 can be inserted in either orientation, include only a partial or one complete and one partial copy of the repeat array and also be inserted with sequence from a third genomic location.
References
- Inaba, H.; Greaves, M.; Mullighan, C.G. Acute lymphoblastic leukaemia. Lancet 2013, 381, 1943–1955.
- Tasian, S.K.; Loh, M.L.; Hunger, S.P. Childhood acute lymphoblastic leukemia: Integrating genomics into therapy. Cancer 2015, 121, 3577–3590.
- Hunger, S.P.; Mullighan, C.G. Redefining ALL classification: toward detecting high-risk ALL and implementing precision medicine. Blood 2015, 125, 3977–3987.
- Harvey, R.C.; Mullighan, C.G.; Chen, I.-M.; Wharton, W.; Mikhail, F.M.; Carroll, A.J.; Kang, H.; Liu, W.; Dobbin, K.K.; Smith, M.A.; et al. Rearrangement of CRLF2 is associated with mutation of JAK kinases, alteration of IKZF1, Hispanic/Latino ethnicity, and a poor outcome in pediatric B-progenitor acute lymphoblastic leukemia. Blood 2010, 115, 5312–5321.
- Mullighan, C.G.; Collins-Underwood, J.R.; Phillips, L.A.A.; Loudin, M.G.; Liu, W.; Zhang, J.; Ma, J.; Coustan-Smith, E.; Harvey, R.C.; Willman, C.L.; et al. Rearrangement of CRLF2 in B-progenitor-and Down syndrome-associated acute lymphoblastic leukemia. Genet. 2009, 41, 1243–1246. doi:10.1038/ng.469.
- Yeoh, E.-J.; Ross, M.E.; Shurtleff, S.A.; Williams, W.K.; Patel, D.; Mahfouz, R.; Behm, F.G.; Raimondi, S.C.; Relling, M.V.; Patel, A.; et al. Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell 2002, 1, 133–143.
- Mullighan, C.G.; Miller, C.B.; Su, X.; Radtke, I.; Dalton, J.; Song, G.; Zhou, X.; Pui, C.-H.; Shurtleff, S.A.; Downing, J.R. ERG Deletions Define a Novel Subtype of B-Progenitor Acute Lymphoblastic Leukemia. Blood 2007, 110, 691–691.
- Harvey, R.C.; Mullighan, C.G.; Wang, X.; Dobbin, K.K.; Davidson, G.S.; Bedrick, E.J.; Chen, I.-M.; Atlas, S.R.; Kang, H.; Ar, K.; et al. Identification of novel cluster groups in pediatric high-risk B-precursor acute lymphoblastic leukemia with gene expression profiling: correlation with genome-wide DNA copy number alterations, clinical characteristics, and outcome. Blood 2010, 116, 4874–4884.
- Zhang, J.; McCastlain, K.; Yoshihara, H.; Xu, B.; Chang, Y.; Churchman, M.L.; Wu, G.; Li, Y.; Wei, L.; Iacobucci, I.; et al. Deregulation of DUX4 and ERG in acute lymphoblastic leukemia. Genet. 2016, 48, 1481–1489.
- Zaliova, M.; Potuckova, E.; Hovorkova, L.; Musilova, A.; Winkowska, L.; Fiser, K.; Stuchly, J.; Mejstrikova, E.; Starkova, J.; Zuna, J.; et al. ERG deletions in childhood acute lymphoblastic leukemia with DUX4 rearrangements are mostly polyclonal, prognostically relevant and their detection rate strongly depends on screening method sensitivity. Haematologica 2019, 104, 1407–1416.
- Potuckova, E.; Zuna, J.; Hovorkova, L.; Starkova, J.; Stary, J.; Trka, J.; Zaliova, M. Intragenic ERG Deletions Do Not Explain the Biology of ERG-Related Acute Lymphoblastic Leukemia. PLoS One 2016, 11, e0160385.
- Yasuda, T.; Tsuzuki, S.; Kawazu, M.; Hayakawa, F.; Kojima, S.; Ueno, T.; Imoto, N.; Kohsaka, S.; Kunita, A.; Doi, K.; et al. Recurrent DUX4 fusions in B cell acute lymphoblastic leukemia of adolescents and young adults. Genet. 2016, 48, 569–574.
- Lilljebjörn, H.; Henningsson, R.; Hyrenius-Wittsten, A.; Olsson, L.; Orsmark-Pietras, C.; von Palffy, S.; Askmyr, M.; Rissler, M.; Schrappe, M.; Cario, G.; et al. Identification of ETV6-RUNX1-like and DUX4-rearranged subtypes in paediatric B-cell precursor acute lymphoblastic leukaemia. Commun. 2016, 7, 11790.
- Zaliova, M.; Stuchly, J.; Winkowska, L.; Musilova, A.; Fiser, K.; Slamova, M.; Starkova, J.; Vaskova, M.; Hrusak, O.; Sramkova, L.; et al. Genomic landscape of pediatric B-other acute lymphoblastic leukemia in a consecutive European cohort. Haematologica 2019, 104, 1396–1406.
- Marincevic-Zuniga, Y.; Dahlberg, J.; Nilsson, S.; Raine, A.; Nystedt, S.; Lindqvist, C.M.; Berglund, E.C.; Abrahamsson, J.; Cavelier, L.; Forestier, E.; et al. Transcriptome sequencing in pediatric acute lymphoblastic leukemia identifies fusion genes associated with distinct DNA methylation profiles. Hematol. Oncol. 2017, 10, 148.
- Vendramini, E.; Giordan, M.; Giarin, E.; Michielotto, B.; Fazio, G.; Cazzaniga, G.; Biondi, A.; Silvestri, D.; Valsecchi, M.G.; Muckenthaler, M.U.; et al. High expression of miR-125b-2 and SNORD116 noncoding RNA clusters characterize ERG-related B cell precursor acute lymphoblastic leukemia. Oncotarget 2017, 8, 42398–42413.
- Gu, Z.; Churchman, M.L.; Roberts, K.G.; Moore, I.; Zhou, X.; Nakitandwe, J.; Hagiwara, K.; Pelletier, S.; Gingras, S.; Berns, H.; et al. PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Genet. 2019, 51, 296–307.
- Li, J.F.; Dai, Y.T.; Lilljebjörn, H.; Shen, S.H.; Cui, B.W.; Bai, L.; Liu, Y.F.; Qian, M.X.; Kubota, Y.; Kiyoi, H.; et al. Transcriptional landscape of B cell precursor acute lymphoblastic leukemia based on an international study of 1,223 cases. Natl. Acad. Sci. U. S. A. 2018, 115, E11711–E11720. doi:10.1073/pnas.1814397115.
- Liu, Y.-F.; Wang, B.-Y.; Zhang, W.-N.; Huang, J.-Y.; Li, B.-S.; Zhang, M.; Jiang, L.; Li, J.-F.; Wang, M.-J.; Dai, Y.-J.; et al. Genomic Profiling of Adult and Pediatric B-cell Acute Lymphoblastic Leukemia. EBioMedicine 2016, 8, 173–183.
- Schroeder, M.P.; Bastian, L.; Eckert, C.; Gökbuget, N.; James, A.R.; Tanchez, J.O.; Schlee, C.; Isaakidis, K.; Häupl, B.; Baum, K.; et al. Integrated analysis of relapsed B-cell precursor Acute Lymphoblastic Leukemia identifies subtype-specific cytokine and metabolic signatures. Rep. 2019, 9, 4188.
- James, A.R.; Schroeder, M.P.; Neumann, M.; Bastian, L.; Eckert, C.; Gökbuget, N.; Tanchez, J.O.; Schlee, C.; Isaakidis, K.; Schwartz, S.; et al. Long non-coding RNAs defining major subtypes of B cell precursor acute lymphoblastic leukemia. Hematol. Oncol. 2019, 12, 8.
- Winokur, S.T.; Bengtsson, U.; Feddersen, J.; Mathews, K.D.; Weiffenbach, B.; Bailey, H.; Markovich, R.P.; Murray, J.C.; Wasmuth, J.J.; Altherr, M.R. The DNA rearrangement associated with facioscapulohumeral muscular dystrophy involves a heterochromatin-associated repetitive element: implications for a role of chromatin structure in the pathogenesis of the disease. Chromosome Res. 1994, 2, 225–234.
- Cacurri, S.; Piazzo, N.; Deidda, G.; Vigneti, E.; Galluzzi, G.; Colantoni, L.; Merico, B.; Ricci, E.; Felicetti, L. Sequence homology between 4qter and 10qter loci facilitates the instability of subtelomeric KpnI repeat units implicated in facioscapulohumeral muscular dystrophy. J. Hum. Genet. 1998, 63, 181–190.
- van Geel, M.; Dickson, M.C.; Beck, A.F.; Bolland, D.J.; Frants, R.R.; van der Maarel, S.M.; de Jong, P.J.; Hewitt, J.E. Genomic analysis of human chromosome 10q and 4q telomeres suggests a common origin. Genomics 2002, 79, 210–217.
- van Deutekom, J.C.; Wijmenga, C.; van Tienhoven, E.A.; Gruter, A.M.; Hewitt, J.E.; Padberg, G.W.; van Ommen, G.J.; Hofker, M.H.; Frants, R.R. FSHD associated DNA rearrangements are due to deletions of integral copies of a 3.2 kb tandemly repeated unit. Mol. Genet. 1993, 2, 2037–2042.
- Geng, L.N.; Yao, Z.; Snider, L.; Fong, A.P.; Cech, J.N.; Young, J.M.; van der Maarel, S.M.; Ruzzo, W.L.; Gentleman, R.C.; Tawil, R.; et al. DUX4 activates germline genes, retroelements, and immune mediators: implications for facioscapulohumeral dystrophy. Cell 2012, 22, 38–51.
- Snider, L.; Geng, L.N.; Lemmers, R.J.L.F.; Kyba, M.; Ware, C.B.; Nelson, A.M.; Tawil, R.; Filippova, G.N.; van der Maarel, S.M.; Tapscott, S.J.; et al. Facioscapulohumeral dystrophy: Incomplete suppression of a retrotransposed gene. PLoS Genet. 2010, 6, e1001181.
- Gabriëls, J.; Beckers, M.C.; Ding, H.; De Vriese, A.; Plaisance, S.; van der Maarel, S.M.; Padberg, G.W.; Frants, R.R.; Hewitt, J.E.; Collen, D.; et al. Nucleotide sequence of the partially deleted D4Z4 locus in a patient with FSHD identifies a putative gene within each 3.3 kb element. Gene 1999, 236, 25–32.
- Choi, S.H.; Gearhart, M.D.; Cui, Z.; Bosnakovski, D.; Kim, M.; Schennum, N.; Kyba, M. DUX4 recruits p300/CBP through its C-terminus and induces global H3K27 acetylation changes. Nucleic Acids Res. 2016, 44, 5161–5173.
- Wijmenga, C.; Hewitt, J.E.; Sandkuijl, L.A.; Clark, L.N.; Wright, T.J.; Dauwerse, H.G.; Gruter, A.M.; Hofker, M.H.; Moerer, P.; Williamson, R. Chromosome 4q DNA rearrangements associated with facioscapulohumeral muscular dystrophy. Genet. 1992, 2, 26–30.
- Pandya, S.; King, W.M.; Tawil, R. Facioscapulohumeral dystrophy. Ther. 2008, 88, 105–113.
- Zeng, W.; de Greef, J.C.; Chen, Y.-Y.; Chien, R.; Kong, X.; Gregson, H.C.; Winokur, S.T.; Pyle, A.; Robertson, K.D.; Schmiesing, J.A.; et al. Specific loss of histone H3 lysine 9 trimethylation and HP1gamma/cohesin binding at D4Z4 repeats is associated with facioscapulohumeral dystrophy (FSHD). PLoS Genet. 2009, 5, e1000559.
- van Overveld, P.G.M.; Lemmers, R.J.F.L.; Sandkuijl, L.A.; Enthoven, L.; Winokur, S.T.; Bakels, F.; Padberg, G.W.; van Ommen, G.-J.B.; Frants, R.R.; van der Maarel, S.M. Hypomethylation of D4Z4 in 4q-linked and non-4q-linked facioscapulohumeral muscular dystrophy. Genet. 2003, 35, 315–317.
- Lemmers, R.J.L.F.; van der Vliet, P.J.; Klooster, R.; Sacconi, S.; Camaño, P.; Dauwerse, J.G.; Snider, L.; Straasheijm, K.R.; van Ommen, G.J.; Padberg, G.W.; et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science 2010, 329, 1650–1653.
- Dmitriev, P.; Kairov, U.; Robert, T.; Barat, A.; Lazar, V.; Carnac, G.; Laoudj-Chenivesse, D.; Vassetzky, Y.S. Cancer-related genes in the transcription signature of facioscapulohumeral dystrophy myoblasts and myotubes. Cell. Mol. Med. 2014, 18, 208–217.
- Dib, C.; Zakharova, V.; Popova, E.; Kiseleva, E.; Chernyak, B.; Lipinski, M.; Vassetzky, Y.S. DUX4 Pathological Expression: Causes and Consequences in Cancer. Trends Cancer Res. 2019, 5, 268–271.
- Kawamura-Saito, M.; Yamazaki, Y.; Kaneko, K.; Kawaguchi, N.; Kanda, H.; Mukai, H.; Gotoh, T.; Motoi, T.; Fukayama, M.; Aburatani, H.; et al. Fusion between CIC and DUX4 up-regulates PEA3 family genes in Ewing-like sarcomas with t(4;19)(q35;q13) translocation. Mol. Genet. 2006, 15, 2125–2137.
- Dyer, M.J.S.; Akasaka, T.; Capasso, M.; Dusanjh, P.; Lee, Y.F.; Karran, E.L.; Nagel, I.; Vater, I.; Cario, G.; Siebert, R. Immunoglobulin heavy chain locus chromosomal translocations in B-cell precursor acute lymphoblastic leukemia: Rare clinical curios or potent genetic drivers? Blood 2010, 115, 1490–1499. doi:1182/blood-2009-09-235986.
- Tian, L.; Shao, Y.; Nance, S.; Dang, J.; Xu, B.; Ma, X.; Li, Y.; Ju, B.; Dong, L.; Newman, S.; et al. Long-read sequencing unveils IGH-DUX4 translocation into the silenced IGH allele in B-cell acute lymphoblastic leukemia. Commun. 2019, 10, 2789.
- Zur Stadt, U.; Alawi, M.; Adao, M.; Indenbirken, D.; Escherich, G.; Horstmann, M.A. Characterization of novel, recurrent genomic rearrangements as sensitive MRD targets in childhood B-cell precursor ALL. Blood Cancer J. 2019, 9, 96.
References
- Inaba, H.; Greaves, M.; Mullighan, C.G. Acute lymphoblastic leukaemia. Lancet 2013, 381, 1943–1955.
- Tasian, S.K.; Loh, M.L.; Hunger, S.P. Childhood acute lymphoblastic leukemia: Integrating genomics into therapy. Cancer 2015, 121, 3577–3590.
- Hunger, S.P.; Mullighan, C.G. Redefining ALL classification: toward detecting high-risk ALL and implementing precision medicine. Blood 2015, 125, 3977–3987.
- Harvey, R.C.; Mullighan, C.G.; Chen, I.-M.; Wharton, W.; Mikhail, F.M.; Carroll, A.J.; Kang, H.; Liu, W.; Dobbin, K.K.; Smith, M.A.; et al. Rearrangement of CRLF2 is associated with mutation of JAK kinases, alteration of IKZF1, Hispanic/Latino ethnicity, and a poor outcome in pediatric B-progenitor acute lymphoblastic leukemia. Blood 2010, 115, 5312–5321.
- Mullighan, C.G.; Collins-Underwood, J.R.; Phillips, L.A.A.; Loudin, M.G.; Liu, W.; Zhang, J.; Ma, J.; Coustan-Smith, E.; Harvey, R.C.; Willman, C.L.; et al. Rearrangement of CRLF2 in B-progenitor-and Down syndrome-associated acute lymphoblastic leukemia. Genet. 2009, 41, 1243–1246. doi:10.1038/ng.469.
- Yeoh, E.-J.; Ross, M.E.; Shurtleff, S.A.; Williams, W.K.; Patel, D.; Mahfouz, R.; Behm, F.G.; Raimondi, S.C.; Relling, M.V.; Patel, A.; et al. Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell 2002, 1, 133–143.
- Mullighan, C.G.; Miller, C.B.; Su, X.; Radtke, I.; Dalton, J.; Song, G.; Zhou, X.; Pui, C.-H.; Shurtleff, S.A.; Downing, J.R. ERG Deletions Define a Novel Subtype of B-Progenitor Acute Lymphoblastic Leukemia. Blood 2007, 110, 691–691.
- Harvey, R.C.; Mullighan, C.G.; Wang, X.; Dobbin, K.K.; Davidson, G.S.; Bedrick, E.J.; Chen, I.-M.; Atlas, S.R.; Kang, H.; Ar, K.; et al. Identification of novel cluster groups in pediatric high-risk B-precursor acute lymphoblastic leukemia with gene expression profiling: correlation with genome-wide DNA copy number alterations, clinical characteristics, and outcome. Blood 2010, 116, 4874–4884.
- Zhang, J.; McCastlain, K.; Yoshihara, H.; Xu, B.; Chang, Y.; Churchman, M.L.; Wu, G.; Li, Y.; Wei, L.; Iacobucci, I.; et al. Deregulation of DUX4 and ERG in acute lymphoblastic leukemia. Genet. 2016, 48, 1481–1489.
- Zaliova, M.; Potuckova, E.; Hovorkova, L.; Musilova, A.; Winkowska, L.; Fiser, K.; Stuchly, J.; Mejstrikova, E.; Starkova, J.; Zuna, J.; et al. ERG deletions in childhood acute lymphoblastic leukemia with DUX4 rearrangements are mostly polyclonal, prognostically relevant and their detection rate strongly depends on screening method sensitivity. Haematologica 2019, 104, 1407–1416.
- Potuckova, E.; Zuna, J.; Hovorkova, L.; Starkova, J.; Stary, J.; Trka, J.; Zaliova, M. Intragenic ERG Deletions Do Not Explain the Biology of ERG-Related Acute Lymphoblastic Leukemia. PLoS One 2016, 11, e0160385.
- Yasuda, T.; Tsuzuki, S.; Kawazu, M.; Hayakawa, F.; Kojima, S.; Ueno, T.; Imoto, N.; Kohsaka, S.; Kunita, A.; Doi, K.; et al. Recurrent DUX4 fusions in B cell acute lymphoblastic leukemia of adolescents and young adults. Genet. 2016, 48, 569–574.
- Lilljebjörn, H.; Henningsson, R.; Hyrenius-Wittsten, A.; Olsson, L.; Orsmark-Pietras, C.; von Palffy, S.; Askmyr, M.; Rissler, M.; Schrappe, M.; Cario, G.; et al. Identification of ETV6-RUNX1-like and DUX4-rearranged subtypes in paediatric B-cell precursor acute lymphoblastic leukaemia. Commun. 2016, 7, 11790.
- Zaliova, M.; Stuchly, J.; Winkowska, L.; Musilova, A.; Fiser, K.; Slamova, M.; Starkova, J.; Vaskova, M.; Hrusak, O.; Sramkova, L.; et al. Genomic landscape of pediatric B-other acute lymphoblastic leukemia in a consecutive European cohort. Haematologica 2019, 104, 1396–1406.
- Marincevic-Zuniga, Y.; Dahlberg, J.; Nilsson, S.; Raine, A.; Nystedt, S.; Lindqvist, C.M.; Berglund, E.C.; Abrahamsson, J.; Cavelier, L.; Forestier, E.; et al. Transcriptome sequencing in pediatric acute lymphoblastic leukemia identifies fusion genes associated with distinct DNA methylation profiles. Hematol. Oncol. 2017, 10, 148.
- Vendramini, E.; Giordan, M.; Giarin, E.; Michielotto, B.; Fazio, G.; Cazzaniga, G.; Biondi, A.; Silvestri, D.; Valsecchi, M.G.; Muckenthaler, M.U.; et al. High expression of miR-125b-2 and SNORD116 noncoding RNA clusters characterize ERG-related B cell precursor acute lymphoblastic leukemia. Oncotarget 2017, 8, 42398–42413.
- Gu, Z.; Churchman, M.L.; Roberts, K.G.; Moore, I.; Zhou, X.; Nakitandwe, J.; Hagiwara, K.; Pelletier, S.; Gingras, S.; Berns, H.; et al. PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Genet. 2019, 51, 296–307.
- Li, J.F.; Dai, Y.T.; Lilljebjörn, H.; Shen, S.H.; Cui, B.W.; Bai, L.; Liu, Y.F.; Qian, M.X.; Kubota, Y.; Kiyoi, H.; et al. Transcriptional landscape of B cell precursor acute lymphoblastic leukemia based on an international study of 1,223 cases. Natl. Acad. Sci. U. S. A. 2018, 115, E11711–E11720. doi:10.1073/pnas.1814397115.
- Liu, Y.-F.; Wang, B.-Y.; Zhang, W.-N.; Huang, J.-Y.; Li, B.-S.; Zhang, M.; Jiang, L.; Li, J.-F.; Wang, M.-J.; Dai, Y.-J.; et al. Genomic Profiling of Adult and Pediatric B-cell Acute Lymphoblastic Leukemia. EBioMedicine 2016, 8, 173–183.
- Schroeder, M.P.; Bastian, L.; Eckert, C.; Gökbuget, N.; James, A.R.; Tanchez, J.O.; Schlee, C.; Isaakidis, K.; Häupl, B.; Baum, K.; et al. Integrated analysis of relapsed B-cell precursor Acute Lymphoblastic Leukemia identifies subtype-specific cytokine and metabolic signatures. Rep. 2019, 9, 4188.
- James, A.R.; Schroeder, M.P.; Neumann, M.; Bastian, L.; Eckert, C.; Gökbuget, N.; Tanchez, J.O.; Schlee, C.; Isaakidis, K.; Schwartz, S.; et al. Long non-coding RNAs defining major subtypes of B cell precursor acute lymphoblastic leukemia. Hematol. Oncol. 2019, 12, 8.
- Winokur, S.T.; Bengtsson, U.; Feddersen, J.; Mathews, K.D.; Weiffenbach, B.; Bailey, H.; Markovich, R.P.; Murray, J.C.; Wasmuth, J.J.; Altherr, M.R. The DNA rearrangement associated with facioscapulohumeral muscular dystrophy involves a heterochromatin-associated repetitive element: implications for a role of chromatin structure in the pathogenesis of the disease. Chromosome Res. 1994, 2, 225–234.
- Cacurri, S.; Piazzo, N.; Deidda, G.; Vigneti, E.; Galluzzi, G.; Colantoni, L.; Merico, B.; Ricci, E.; Felicetti, L. Sequence homology between 4qter and 10qter loci facilitates the instability of subtelomeric KpnI repeat units implicated in facioscapulohumeral muscular dystrophy. J. Hum. Genet. 1998, 63, 181–190.
- van Geel, M.; Dickson, M.C.; Beck, A.F.; Bolland, D.J.; Frants, R.R.; van der Maarel, S.M.; de Jong, P.J.; Hewitt, J.E. Genomic analysis of human chromosome 10q and 4q telomeres suggests a common origin. Genomics 2002, 79, 210–217.
- van Deutekom, J.C.; Wijmenga, C.; van Tienhoven, E.A.; Gruter, A.M.; Hewitt, J.E.; Padberg, G.W.; van Ommen, G.J.; Hofker, M.H.; Frants, R.R. FSHD associated DNA rearrangements are due to deletions of integral copies of a 3.2 kb tandemly repeated unit. Mol. Genet. 1993, 2, 2037–2042.
- Geng, L.N.; Yao, Z.; Snider, L.; Fong, A.P.; Cech, J.N.; Young, J.M.; van der Maarel, S.M.; Ruzzo, W.L.; Gentleman, R.C.; Tawil, R.; et al. DUX4 activates germline genes, retroelements, and immune mediators: implications for facioscapulohumeral dystrophy. Cell 2012, 22, 38–51.
- Snider, L.; Geng, L.N.; Lemmers, R.J.L.F.; Kyba, M.; Ware, C.B.; Nelson, A.M.; Tawil, R.; Filippova, G.N.; van der Maarel, S.M.; Tapscott, S.J.; et al. Facioscapulohumeral dystrophy: Incomplete suppression of a retrotransposed gene. PLoS Genet. 2010, 6, e1001181.
- Gabriëls, J.; Beckers, M.C.; Ding, H.; De Vriese, A.; Plaisance, S.; van der Maarel, S.M.; Padberg, G.W.; Frants, R.R.; Hewitt, J.E.; Collen, D.; et al. Nucleotide sequence of the partially deleted D4Z4 locus in a patient with FSHD identifies a putative gene within each 3.3 kb element. Gene 1999, 236, 25–32.
- Choi, S.H.; Gearhart, M.D.; Cui, Z.; Bosnakovski, D.; Kim, M.; Schennum, N.; Kyba, M. DUX4 recruits p300/CBP through its C-terminus and induces global H3K27 acetylation changes. Nucleic Acids Res. 2016, 44, 5161–5173.
- Wijmenga, C.; Hewitt, J.E.; Sandkuijl, L.A.; Clark, L.N.; Wright, T.J.; Dauwerse, H.G.; Gruter, A.M.; Hofker, M.H.; Moerer, P.; Williamson, R. Chromosome 4q DNA rearrangements associated with facioscapulohumeral muscular dystrophy. Genet. 1992, 2, 26–30.
- Pandya, S.; King, W.M.; Tawil, R. Facioscapulohumeral dystrophy. Ther. 2008, 88, 105–113.
- Zeng, W.; de Greef, J.C.; Chen, Y.-Y.; Chien, R.; Kong, X.; Gregson, H.C.; Winokur, S.T.; Pyle, A.; Robertson, K.D.; Schmiesing, J.A.; et al. Specific loss of histone H3 lysine 9 trimethylation and HP1gamma/cohesin binding at D4Z4 repeats is associated with facioscapulohumeral dystrophy (FSHD). PLoS Genet. 2009, 5, e1000559.
- van Overveld, P.G.M.; Lemmers, R.J.F.L.; Sandkuijl, L.A.; Enthoven, L.; Winokur, S.T.; Bakels, F.; Padberg, G.W.; van Ommen, G.-J.B.; Frants, R.R.; van der Maarel, S.M. Hypomethylation of D4Z4 in 4q-linked and non-4q-linked facioscapulohumeral muscular dystrophy. Genet. 2003, 35, 315–317.
- Lemmers, R.J.L.F.; van der Vliet, P.J.; Klooster, R.; Sacconi, S.; Camaño, P.; Dauwerse, J.G.; Snider, L.; Straasheijm, K.R.; van Ommen, G.J.; Padberg, G.W.; et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science 2010, 329, 1650–1653.
- Dmitriev, P.; Kairov, U.; Robert, T.; Barat, A.; Lazar, V.; Carnac, G.; Laoudj-Chenivesse, D.; Vassetzky, Y.S. Cancer-related genes in the transcription signature of facioscapulohumeral dystrophy myoblasts and myotubes. Cell. Mol. Med. 2014, 18, 208–217.
- Dib, C.; Zakharova, V.; Popova, E.; Kiseleva, E.; Chernyak, B.; Lipinski, M.; Vassetzky, Y.S. DUX4 Pathological Expression: Causes and Consequences in Cancer. Trends Cancer Res. 2019, 5, 268–271.
- Kawamura-Saito, M.; Yamazaki, Y.; Kaneko, K.; Kawaguchi, N.; Kanda, H.; Mukai, H.; Gotoh, T.; Motoi, T.; Fukayama, M.; Aburatani, H.; et al. Fusion between CIC and DUX4 up-regulates PEA3 family genes in Ewing-like sarcomas with t(4;19)(q35;q13) translocation. Mol. Genet. 2006, 15, 2125–2137.
- Dyer, M.J.S.; Akasaka, T.; Capasso, M.; Dusanjh, P.; Lee, Y.F.; Karran, E.L.; Nagel, I.; Vater, I.; Cario, G.; Siebert, R. Immunoglobulin heavy chain locus chromosomal translocations in B-cell precursor acute lymphoblastic leukemia: Rare clinical curios or potent genetic drivers? Blood 2010, 115, 1490–1499. doi:1182/blood-2009-09-235986.
- Tian, L.; Shao, Y.; Nance, S.; Dang, J.; Xu, B.; Ma, X.; Li, Y.; Ju, B.; Dong, L.; Newman, S.; et al. Long-read sequencing unveils IGH-DUX4 translocation into the silenced IGH allele in B-cell acute lymphoblastic leukemia. Commun. 2019, 10, 2789.
- Zur Stadt, U.; Alawi, M.; Adao, M.; Indenbirken, D.; Escherich, G.; Horstmann, M.A. Characterization of novel, recurrent genomic rearrangements as sensitive MRD targets in childhood B-cell precursor ALL. Blood Cancer J. 2019, 9, 96.