Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Peter Tang and Version 1 by Zsolt Gáll.
Vitamin D is necessary for all vertebrates, including humans, to maintain adequate phosphate and calcium levels in the blood, thereby helping to develop normal bone, optimal maintenance of muscle contractions, and cellular functions in different parts of the body. The developmental disabilities induced by vitamin D deficiency (VDD) include neurological disorders (e.g., attention deficit hyperactivity disorder, autism spectrum disorder, schizophrenia) characterized by cognitive dysfunction.
- vitamin D
- vitamin D deficiency
- central nervous system
- cognitive function
- brain development
1. Introduction
Vitamin D deficiency (VDD) affects almost one billion people today [1]. It is now recognized that VDD can be responsible for the development of several extraskeletal pathologies, in addition to rickets and osteomalacia, such as multiple sclerosis, autoimmune disorders, the incidence of infections, respiratory diseases, cardiovascular disorders, and various types of cancers [1]. The development of VDD poses a particular risk to individuals with limited intake, insufficient exposure to sunlight, or inadequate intestinal absorption [2]. Vitamin D3 is synthesized in the skin by ultraviolet (UV) radiation. Air pollution, a common problem in developing countries, can also contribute to the development of VDD, as particulate matter absorbs UVB radiation or reflects sunlight [3]. At the same time, the habit of dressing may play a role in developing VDD since the synthesis of vitamin D in the skin is hampered [4]. Decreased 7-dehydrocholesterol (7-DHC) in the epidermis, inadequate consumption of certain foods such as dairy products, and impaired vitamin D metabolism in renal and hepatic insufficiency can also lead to deficiency [5]. On the other hand, certain drugs (e.g., immunosuppressants, glucocorticoids, antiepileptics, antifungals, and antiretrovirals) may cause increased catabolism of vitamin D resulting in VDD [1,5,6][1][5][6].
The term “vitamin D” is a general one that refers to several structurally related secosteroids, including cholecalciferol, ergocalciferol, 25-hydroxyvitamin D (calcidiol), or for 1,25-dihydroxyvitamin D (calcitriol) [7]. For the sake of clarity, in this reviewere, the term vitamin D is used to refer to the animal-derived cholecalciferol, or vitamin D3, and to the plant-derived ergocalciferol, or vitamin D2, except when discussing VDD, which is defined based on serum calcidiol levels. All of them are biologically inactive and need activation through a series of enzymatic transformations. The active forms of both compounds (i.e., 1,25-dihydroxy vitamin D3 and 1,25-dihydroxy vitamin D2) are pleiotropic secosteroid hormones, being involved in regulating the expression of more than 900 genes by binding to the vitamin D receptor (VDR, NR1|1) that acts as a nuclear transcription factor after the formation of a VDR/retinoid-X receptor/cofactor complex.
Vitamin D is necessary for all vertebrates, including humans, to maintain adequate phosphate and calcium levels in the blood, thereby helping to develop normal bone, optimal maintenance of muscle contractions, and cellular functions in different parts of the body [8]. Along with the best-known effects of regulating calcium absorption and promoting bone mineralization, the target genes are involved with a wide variety of biological processes, such as cell proliferation and differentiation, immune response and cytokine regulation [9]. In the last two decades, convincing evidence has been published that indicate the implication of the vitamin D signaling system in several brain functions and disorders.
2. Molecular Evidence of the Relationship between Vitamin D and Cognitive Function
Vitamin D can exert its effect on neurocognition through a number of mechanisms such as induction of neuroprotection, modulation of oxidative stress, regulation of calcium homeostasis, and inhibition of inflammatory processes [10]. Vitamin D acts through its own receptor, VDR, a nuclear hormone receptor found in the central nervous system. VDR has a DNA-binding domain containing two zinc fingers: one of the regions is responsible for DNA binding, and the other is involved in the dimerization of the molecule. The ligand-binding domain at the C-terminal end of the receptor ensures the specificity and selectivity of the response. There is ample evidence that vitamin D exerts a genomic effect on the brain. VDR, found in both developing and adult brains in rodents, has been shown to be present in the neuroepithelium and midbrain of a 12-day-old embryo (E12) [13][11]. It is present in neurons, glia cells, especially in the temporal, cingulate, and orbital cortex, thalamus, nucleus accumbens, terminal stretch marks, and amygdala, all of which are essential for the development of cognitive functions [14][12]. It is also expressed in the CA1, CA2, CA3, and CA4 layers of hippocampal pyramidal cells in both rodent and human brains [15][13]. The distribution of VDR in humans is strikingly similar to that observed in rodents. Its presence in the hippocampus, cerebral cortex, and limbic system of humans and rodents reinforces the role of vitamin D in regulating learning and memory, however, VDR is also localized in the olfactory, visual, and auditory systems, so it may also play a role in somatosensory functions, which could contribute to better cognitive task performances [14,15][12][13]. Furthermore, the diverse distribution of VDR in the brain suggests that vitamin D may be involved in neuronal proliferation and stem-cell differentiation. Indirect evidence for the neurodevelopmental role of vitamin D is represented by the morphological changes that occur in offspring of rats subjected to VDD. Eyles and collaborators created the first dietary developmental VDD model, which was characterized by increased brain cell proliferation [16][14]. On the other hand, developmental VDD has also been demonstrated to affect the expression of genes regulating apoptosis and the cell cycle in rat embryos [17,18][15][16]. In a recent study, neuroprotective effects of cholecalciferol have been shown in young rats [19][17]. Analyzing the development of the whole brain, they described an increase in the volume of lateral ventricles accompanied by a decrease in the hippocampal volume [19][17]. This finding has not been confirmed in adult mice nor in rats [20[18][19],21], but recently it has been described in patients with mild cognitive impairment [22][20]. In another study, VDD has also been associated with a 28% increase in lateral ventricles in aged humans [23][21]. On the molecular level, developmental VDD has been proven to affect brain gene and protein expression in the long term. The altered expression of 74 genes was identified, which were supposed to be involved in various neuronal functions [24][22]. In a proteomic study, 36 proteins involved in calcium homeostasis, neurotransmission, synaptic plasticity, redox balance, oxidative phosphorylation, etc., were dysregulated in the prefrontal cortex and hippocampus of adult animals [25][23]. VDD has been linked with the dysregulation of dopamine and serotonin neurotransmission. In developing brains, the VDR appears at E12, exactly when the dopaminergic system starts to develop [26,27,28][24][25][26]. Some evidence has demonstrated that VDD affects components of dopaminergic neuron-maturation factors (e.g., reduction in neurotrophin brain-derived neurotrophic factor, TGF-β1 [29][27], Nurr1 [30][28] and metabolizing enzymes (e.g., decrease in the expression of catechol-O-methyltransferase [31][29] and tyrosine hydroxylase [29][27]). Moreover, Cui et al. demonstrated that calcitriol increased N-cadherin and tyrosine hydroxylase expression in VDR-expressing neuroblastomas, and importantly, VDD reduced their level in embryonic mesencephalon [26,32][24][30]. These findings strongly suggest that calcitriol triggers the differentiation of dopaminergic neurons [13][11]. Serotonin plays a crucial role in brain development because it is a key modulator of neuronal cell proliferation, migration and brain wiring during fetal and early postnatal life [33][31]. Serotonin turnover may also be influenced due to the regulation of the expression of tryptophan hydroxylase-2 and leptin genes [34,35][32][33]. It was found that after short-term exposure to calcitriol, TPH2 expression increased in cultured serotonergic B14 derived from the rat brain [36][34]. The same study showed that several human-derived cell lines responded to calcitriol treatment in a similar way [36][34]. Moreover, Jiang et al. have performed a complex study to provide direct evidence to the above-mentioned links between calcitriol–dopamine and calcitriol–serotonin in the brain [37][35]. They found that chronic calcitriol administration increased the level of γ-aminobutyric acid (GABA), glutamate, but not that of the dopamine and serotonin in the prefrontal cortex and hippocampus. However, dopamine- and serotonin-turnover were increased, which was demonstrated by the high level of 3,4-dihydroxyphenyl acetic acid (DOPAC) and homovanillic acid (HVA) and 5-hydroxyindole acetic acid (5-HIAA), the metabolites of dopamine and serotonin, respectively. On the other hand, they also showed that high expression of the metabolizing enzymes (i.e., catechol-O-methyltransferase for dopamine and monoamine oxidase A for serotonin) could be responsible for the rapid degradation of the neurotransmitter [37][35]. All the above-mentioned effects of vitamin D are linked to its binding to VDR. However, recently, membrane receptors activated by calcitriol were also identified. The activation of the 1,25D3 membrane-associated, rapid-response steroid-binding protein (1,25D3-MARRS), which also serves as a protein disulfide isomerase A3 (PDIA3), provides a rapid cellular response. PDIA3 possesses several important functions, one of which is to modulate inflammation, apoptosis, and oxidative stress [38][36]. In rats, the expression of Pdia3 mRNA is higher in neurons, astrocytes, and endothelial cells compared to the kidney and liver, so they proposed that Pdia3 is the main vitamin D receptor in the rat brain [39][37]. Early studies in humans suggested that PDIA3 possessed neuroprotective actions against infections or toxic drugs [40,41][38][39]. Moreover, exogenous PDIA3 increased the expression of brain-derived neurotrophic factor (BDNF) and phosphorylated cAMP-response element-binding protein (pCREB), thus enhancing cell proliferation in the hippocampus under normal conditions, but it failed to reduce ischemic alterations in gerbils [42][40]. All these studies supported the hypothesis that activating 1,25D3-MARRS could be involved in calcitriol’s actions. However, surprisingly, the studies conducted on PDIA3 knockout mice reported an attenuated inflammatory response to traumatic brain injury [43][41]. So, the role of PDIA3 in the cognitive processes is still uncertain, and based on these new results, further research on PDIA3 could be reasonable. Moreover, PDIA3 expression has been proposed as a biomarker in several types of cancer, and increased expression promotes proinflammatory cytokine release [44][42].3. Transport and Cellular Uptake of Vitamin D
A fully functional brain signaling system involves the presence of ligands in adequate quantity at the site of action. Vitamin D and its metabolites are lipophilic compounds assumed to penetrate the cell membranes and the blood–brain barrier (BBB) by diffusion. According to the free hormone hypothesis, only the unbound fraction (or the free fraction) of total vitamin D metabolites found in blood or extracellular fluid can diffuse into the intracellular space and couple to its receptors and exert its effects. However, all vitamin D metabolites bind to DBP in a very high proportion (~99%) resulting in picomolar concentrations of free fraction [45][43]. DBP is synthesized in the liver and circulates in high amounts in the serum, but it can also be found in the interstitial space of various organs. DBP is a low molecular-weight protein, smaller than albumin, but it is essential to build a pool of circulating calcidiol, thus preventing the rapid onset of deficiency. On the other hand, DBP-bound vitamin D metabolites are filtered in the renal glomerulus, but these can be reabsorbed by the proximal tubule through endocytosis by the megalin/cubilin complex [46][44]. Megalin is expressed along with cubilin mainly in the kidney, brain, and eyes. The complex surely has a role in normal brain development, because the decline of its expression is associated with neurodegenerative processes (e.g., Alzheimer’s disease) [47][45]. Theoretically, megalin can bind and internalize DBP-bound calcidiol, thus facilitating the access of megalin-expressing cells to calcidiol, but the existence of such a transport mechanism has not been demonstrated yet [45][43]. Therefore, it can be assumed that the free hormone model is applicable and relevant for calcitriol also. Another important piece of evidence endorsing vitamin D actions in the brain was the detection of vitamin D metabolites in rodents and the human brain [48,49][46][47]. Early autoradiographic studies demonstrated that calcitriol penetrates the BBB and is distributed in the brain [50][48], but until recently, the quantification of endogenous calcidiol or calcitriol has not been performed from brain tissue. Xue et al. described a correlation between serum and brain total calcidiol concentrations in rats, however, the results should be interpreted with caution, considering that no correction was applied to residual blood from cerebral vessels [48][46]. It is also noteworthy that DBP is found in cerebrospinal fluid (CSF), so vitamin D metabolites are likely to bind to this. However, the expression of DBP in the CNS is influenced by different pathological conditions (e.g., multiple sclerosis, meningitis) [51][49], so the free fractions of calcidiol and calcitriol may fluctuate and are very difficult to quantify.4. Assessment of the Vitamin D Status
In the human body, the main metabolic markers used for the assessment of vitamin D status are hydroxylated metabolites, 25-OHD3 and 25-OHD2, due to their long half-lives (2–3 weeks) and stability [52][50]. In addition, 25-OHD represents the sum of vitamin D intake and dermal synthesis. The current guidelines suggest that total serum 25-OHD is an appropriate biological marker for assessing vitamin D status, so it can be used to determine vitamin deficiency or sufficient or even excessive vitamin levels [53][51]. Deficiency may be indicated by total serum levels of <20 ng/mL (<50 nmol/L) for 25-OHD and total serum levels of 25-OHD between 21–29 ng/mL (52.5–72.5 nmol/L) may be considered insufficient without obvious clinical symptoms [1,15][1][13]. Vitamin D supplementation is the most widely used strategy to restore vitamin deficiency. The recommended intake is 800 IU to reach 25 ng/mL total serum 25-OHD, 1600 IU vitamin D to 30 ng/mL total serum 25-OHD. The estimated average requirement is 400 IU to reach 20 ng/mL and 800 IU to total serum levels of 30 ng/mL [54][52]. The serum is the most important medium for the detection of vitamin D metabolites. Calcidiol is the major circulating form of vitamin D in serum. Serum calcidiol levels provide a significant response to changes in vitamin levels during both sunlight and seasonal changes, as well as in vitamin D supplementation [53][51]. However, the relationship between serum and tissue, especially brain tissue concentrations, is not clarified yet. Furthermore, there is a lack of evidence on the influence of supplementation on brain tissue levels in humans. In rodents, Xue et al. described that the concentration of vitamin D3 metabolites increased in the brain of rats fed with a vitamin D3-rich diet, and they observed that serum and brain levels correlated [48][46]. However, they quantified only the 25-OHD3 and 24,25-OH2-D3 concentrations, the modification of calcitriol concentration in the brain after vitamin D3 supplementation remains controversial. One of the challenges with measuring the concentration of calcitriol in brain tissue is its very low level and a lack of quantitation methods that can determine picomolar concentrations. Still, Fu et al. developed a highly sensitive analytical method and detected calcitriol in some regions of the human postmortem brain along with calcifediol [49][47]. This was the first direct evidence that calcifediol is the most abundant vitamin D3 metabolite not only in the serum but also in the human brain. Cholecalciferol was not detectable, but only one human brain specimen was analyzed, and it was likely from an older subject. So, there is still a need for further investigations on whether cholecalciferol supplementation increases the vitamin D signaling pathway in the brain in order to assess the role of supplementation in certain pathologies. It is important to note that in all available studies, including preclinical and clinical data, VDD was defined based on total serum levels of 25-OHD.5. Synthesis and Catabolism of Calcitriol in the Brain
The activation of vitamin D receptors (VDR or PDIA3) in different brain regions supposes that calcitriol reaches certain concentrations in the interstitial or intracellular space. As presented above, this can happen if calcitriol passes the BBB and gets access to cells. According to the current knowledge, the synthesis of calcitriol takes place through the classical route that begins in the epithelial layers of the intestine, as cholesterol from food or bile is oxidized to 7-DHC, which is then further activated in the liver and kidney (Figure 1) [55][53]. Recently, the hypothesis of augmenting the “systemic” calcitriol action by local production has recently been broadened to include the central nervous system [15][13]. However, the only evidence supporting this is the expression of the enzymes involved in synthesis and catabolism of calcitriol by neurons, glial cells, and microglia. There is no direct evidence for local production or catabolism of calcitriol in different types of brain cells. In this chapter, the expression of the key enzymes will be discussed, highlighting the latest results regarding their expression in the brain.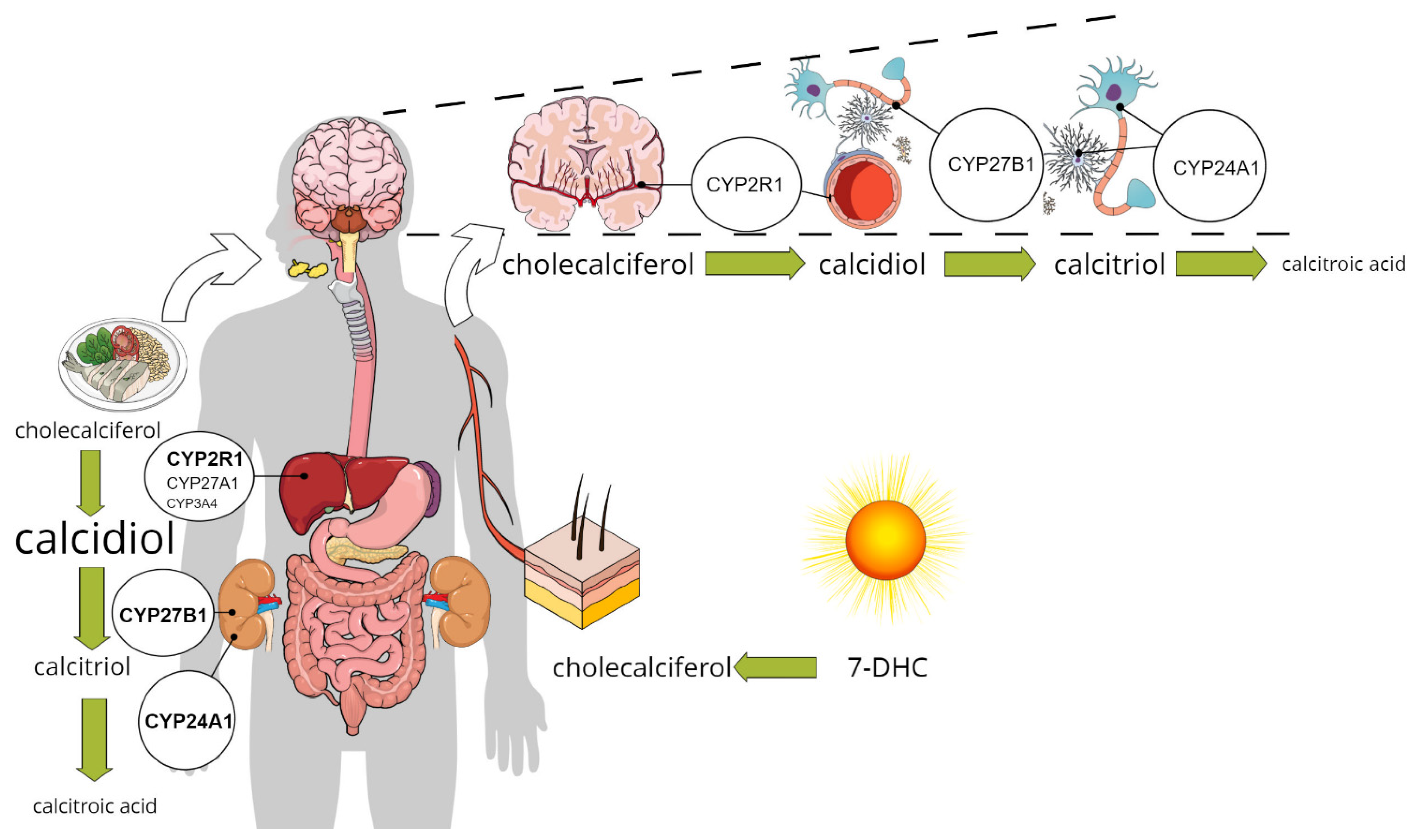
Figure 1. The classical and the alternative pathway for calcitriol synthesis: enzymes involved in calcitriol synthesis are expressed in pericytes, glial cells, and neurons in addition to the liver and kidney, pointing to the possible role for the local production in vitamin D signalling.
6. Influence of Vitamin D3 Deficiency/Supplementation on Cognitive Impairment in Animal Models
The behavioral effects of manipulating vitamin D signaling on brain development have been studied extensively by using genetic knockout (VDR−/− or CYP27B1−/−) models and models of dietary deficiency. Dietary deficiency models might seem to have more construct validity but are time-consuming, labor-intensive, and can be limited in result interpretation. The behavior of rats and mice subjected to dietary developmental VDD has been observed in various experimental conditions. In rats, developmental VDD induced disrupted latent inhibition, which reflects the attentional processing of animals [78][74] but normal prepulse inhibition and working memory. However, these animals showed hyperlocomotion and increased exploratory activity, which might enhance some types of cognitive functions or could be a confounding factor in several behavioral tests used to assess cognitive function [78,84][74][75]. Indeed, electrophysiological studies demonstrated an enhanced long-term potentiation in the hippocampus and better retention performance in the Y-maze test in prenatal VDD animals, both results supporting an improved memory formation in rats [82,85][76][77]. Conversely, mice exhibited learning deficits, although they showed the same increase in exploration and hyperlocomotion as rats [77,80,86][78][79][80]. This means that developmental VDD might influence the various components of cognition differently. Using complex behavioral paradigms, such as the five-choice serial reaction time task (5C-SRT) and the five-choice continuous performance test (5C-CPT), which can reflect the sustained attention and vigilance in rodents, VDD rats had normal performance to target stimuli and maintained high levels of accuracy but had increased probability of false-alarm responding to nonsignal stimuli, and they showed poorer vigilance and a lower responsivity index [87][81]. In mice, VDD mice showed similar results to controls in the primary measures of performance but tended to make more premature responses than control mice [80][79]. These subtle alterations observed in rats and mice may represent a symptom of a compulsive or impulsive behavior rather than cognitive dysfunction. Recently, Overeem et al. described altered recognition memory associated with developmental VDD [79][82], however, the results obtained in the novel object recognition test need further confirmation as this test has a wide range of manipulations, and it also has its limitations [88][83]. Overall, the cognitive impairments reported in the developmental VDD model may be associated with psychiatric disorders such as schizophrenia, autism, or attention deficit hyperactivity disorder, particularly because some of them can be treated with different antipsychotics [84,85,87][75][77][81]. Besides developmental VDD models, several genetic mice models were obtained by VDR gene ablation or mutation in mice [89,90,91][84][85][86]. These animals showed increased levels of PTH and calcitriol along with hypocalcemia, hypophosphatemia, and consequential skeletal disorders [92][87]. Therefore, to study the abnormalities induced by VDR ablation only and not the consequences of altered mineral homeostasis, a calcium- and phosphate-rich diet, a so called “rescue diet”, should be delivered to VDR knockout animals [90,92,93,94][85][87][88][89]. Even so, a great variation in PTH values (from normal to excessive PTH) has been observed along with VDR-independent compensatory mechanisms [95,96][90][91]. On the other hand, increased dietary calcium load induces FGF23 synthesis [97][92], which was shown to affect spatial learning and memory [98][93]. Thus, the contradictory results reported so far between dietary and genetic VDD models may be due to several confounding factors, one of which is the FGF23 [99][94]. Taking into account the above-mentioned endocrinological modifications induced by VDD knockout, the altered behavioral responses are difficult to understand. Burne et al. and Kalueff et al. studied thoroughly the behavioral anomalies caused by the lack of functional VDR in mice and reported an array of neuropsychiatric deficits (e.g., anxiety, neophobia, altered maternal behavior, impaired motor coordination), along with muscle weakness, impaired energy metabolism, and altered cardiac functions [100,101,102,103,104,105,106][95][96][97][98][99][100][101]. As all these alterations affect the performance of mice in a behavioral test, the VDR knockout model is not suitable to study the impact of calcitriol on cognition. Various preclinical models were proposed to study the influence of transient VDD and vitamin D3 supplementation on adults. Hypocalcemia and secondary hyperparathyroidism consecutive to VDD induction have been addressed in different ways by the research groups. Burne’s group applied a vitamin D-free diet for six weeks in rats and ten weeks in different strains of mice and demonstrated an up to a 10-fold decrease in the serum calcidiol levels, below 10 nM in each experiment, associated with normocalcemia [107][102]. However, they did not modify the calcium or phosphorus intake of the animals and did not monitor the serum PTH levels. Nevertheless, they found that this protocol is not sufficient to induce cognitive alterations in the young adult rat, observing a slight effect on vigilance only [107][102]. On the other hand, in ten-week-old mice, spontaneous hyperlocomotion was described similarly to developmental VDD models, but there was no influence of the diet on cognitive functions. Neither the old mice nor the middle-aged rats showed cognitive impairment, even though they had been deprived of vitamin D for more extended periods of time (6–12 months) [108,109][103][104]. One specific learning task did appear to be influenced by VDD in mice, namely the hippocampal-dependent spatial learning. VDD mice exhibited impairments in the active place-avoidance test, which were not linked to motor coordination problems or muscle weakness. It is important to note that connectivity deficits were also observed in the hippocampus [59][57]. Latimer et al. reported that in middle-aged rats subjected to 6-months of vitamin D manipulation through diet, serum calcidiol levels correlated with the performance of the animals in the Morris water maze task, and supraphysiological doses may be protective against mild cognitive impairment [110][105]. Liang et al. performed a similarly designed study in mice, including a group with a supraphysiological dose of vitamin D. In their study, mice were fed life-long with a specific diet containing a higher level of calcium before they were behaviorally assessed at 6-weeks and 17-weeks of age [111][106]. They concluded that altered vitamin D intake had a significant impact on long-term memory and promoted memory loss (not only VDD but also the overdose were harmful) [111][106]. In summary, animal models of VDD have shown that low total serum calcidiol induces behavioral changes, suggesting that calcitriol is involved in a number of important neuronal and glial processes. Interestingly, VDD did not cause cognitive impairment in rodents as would be expected, but vitamin D supplementation improved some of their cognitive processes (Figure 2). Possible explanations for the controversial finding may be related to study design (i.e., animal model, diet, dosing, paraclinical tests used to confirm VDD), but some unknown aspects of vitamin D metabolism in the brain may also play a role.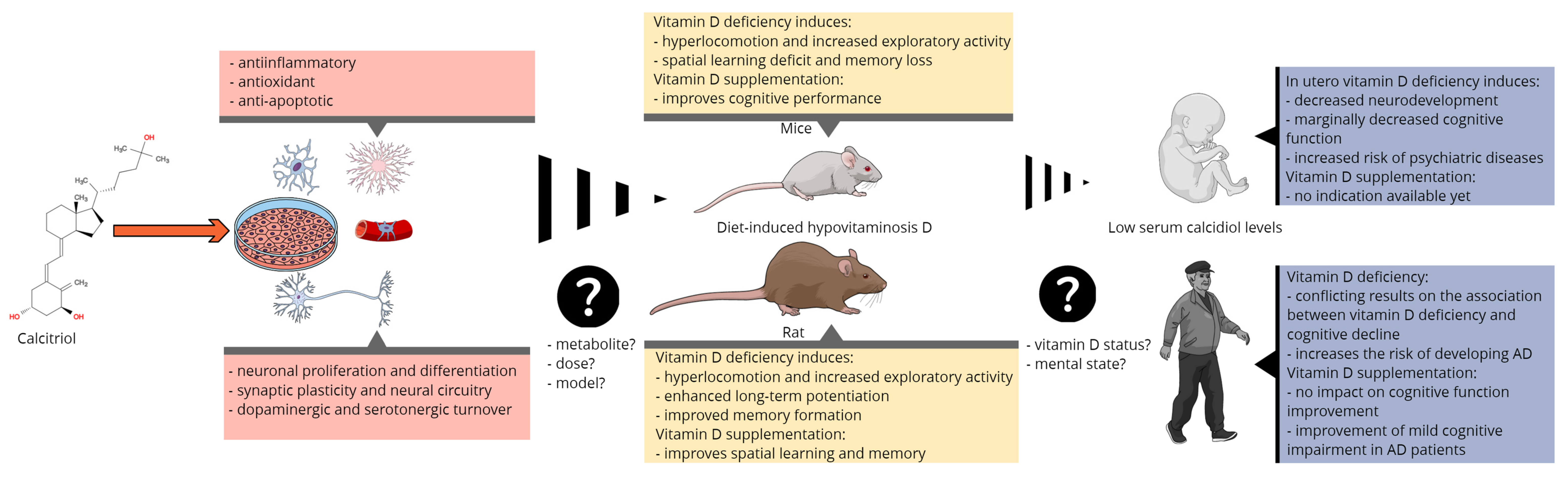
Figure 2.
Summary of the main molecular, preclinical, and clinical findings linking vitamin D to cognitive function and the most important issues of translational research.
References
- Pfotenhauer, K.M.; Shubrook, J.H. Vitamin D deficiency, its role in health and disease, and current supplementation recommendations. J. Am. Osteopath. Assoc. 2017, 117, 301–305.
- Kennel, K.A.; Drake, M.T.; Hurley, D.L. Vitamin D Deficiency in Adults: When to Test and How to Treat. Mayo Clin. Proc. 2010, 85, 752–758.
- Mousavi, S.E.; Amini, H.; Heydarpour, P.; Chermahini, F.A.; Godderis, L. Air pollution, environmental chemicals, and smoking may trigger vitamin D deficiency: Evidence and potential mechanisms. Environ. Int. 2019, 122, 67–90.
- Jarrett, P.; Scragg, R. Evolution, prehistory and vitamin D. Int. J. Environ. Res. Public Health 2020, 17, 646.
- Sahota, O. Understanding vitamin D deficiency. Age Ageing 2014, 43, 589–591.
- Kupisz-Urbańska, M.; Płudowski, P.; Marcinowska-Suchowierska, E. Vitamin d deficiency in older patients—problems of sarcopenia, drug interactions, management in deficiency. Nutrients 2021, 13, 1247.
- Vieth, R. Vitamin D supplementation: Cholecalciferol, calcifediol, and calcitriol. Eur. J. Clin. Nutr. 2020, 74, 1493–1497.
- Jones, G. The discovery and synthesis of the nutritional factor vitamin D. Int. J. Paleopathol. 2018, 23, 96–99.
- Pike, J.W.; Christakos, S. Biology and Mechanisms of Action of the Vitamin D Hormone. Endocrinol. Metab. Clin. N. Am. 2017, 46, 815–843.
- Bivona, G.; Gambino, C.M.; Iacolino, G.; Ciaccio, M. Vitamin D and the nervous system. Neurol. Res. 2019, 41, 827–835.
- Cui, X.; Gooch, H.; Petty, A.; McGrath, J.J.; Eyles, D. Vitamin D and the brain: Genomic and non-genomic actions. Mol. Cell. Endocrinol. 2017, 453, 131–143.
- Eyles, D.W.; Smith, S.; Kinobe, R.; Hewison, M.; McGrath, J.J. Distribution of the Vitamin D receptor and 1α-hydroxylase in human brain. J. Chem. Neuroanat. 2005, 29, 21–30.
- Landel, V.; Annweiler, C.; Millet, P.; Morello, M.; Féron, F. Vitamin D, Cognition and Alzheimer’s Disease: The Therapeutic Benefit is in the D-Tails. J. Alzheimer’s Dis. 2016, 53, 419–444.
- Cui, X.; McGrath, J.J.; Burne, T.H.J.; Mackay-Sim, A.; Eyles, D.W. Maternal vitamin D depletion alters neurogenesis in the developing rat brain. Int. J. Dev. Neurosci. 2007, 25, 227–232.
- Ko, P.; Burkert, R.; McGrath, J.; Eyles, D. Maternal vitamin D3 deprivation and the regulation of apoptosis and cell cycle during rat brain development. Dev. Brain Res. 2004, 153, 61–68.
- Eyles, D.; Brown, J.; Mackay-Sim, A.; McGrath, J.; Feron, F. Vitamin d3 and brain development. Neuroscience 2003, 118, 641–653.
- Şahin, S.; Gürgen, S.G.; Yazar, U.; İnce, İ.; Kamaşak, T.; Arslan, E.A.; Durgut, B.D.; Dilber, B.; Cansu, A. Vitamin D protects against hippocampal apoptosis related with seizures induced by kainic acid and pentylenetetrazol in rats. Epilepsy Res. 2019, 149, 107–116.
- Lardner, A.L. Vitamin D and hippocampal development-the story so far. Front. Mol. Neurosci. 2015, 8, 58.
- Al-Amin, M.M.; Sullivan, R.K.P.; Kurniawan, N.D.; Burne, T.H.J. Adult vitamin D deficiency disrupts hippocampal-dependent learning and structural brain connectivity in BALB/c mice. Brain Struct. Funct. 2019, 224, 1315–1329.
- Al-Amin, M.; Bradford, D.; Sullivan, R.K.P.; Kurniawan, N.D.; Moon, Y.; Han, S.-H.; Zalesky, A.; Burne, T.H.J. Vitamin D deficiency is associated with reduced hippocampal volume and disrupted structural connectivity in patients with mild cognitive impairment. Hum. Brain Mapp. 2019, 40, 394–406.
- Annweiler, C.; Montero-Odasso, M.; Hachinski, V.; Seshadri, S.; Bartha, R.; Beauchet, O. Vitamin D concentration and lateral cerebral ventricle volume in older adults. Mol. Nutr. Food Res. 2013, 57, 267–276.
- Eyles, D.; Almeras, L.; Benech, P.; Patatian, A.; Mackay-Sim, A.; McGrath, J.; Féron, F. Developmental vitamin D deficiency alters the expression of genes encoding mitochondrial, cytoskeletal and synaptic proteins in the adult rat brain. J. Steroid Biochem. Mol. Biol. 2007, 103, 538–545.
- Almeras, L.; Eyles, D.; Benech, P.; Laffite, D.; Villard, C.; Patatian, A.; Boucraut, J.; Mackay-Sim, A.; McGrath, J.; Féron, F. Developmental vitamin D deficiency alters brain protein expression in the adult rat: Implications for neuropsychiatric disorders. Proteomics 2007, 7, 769–780.
- Pertile, R.A.N.; Cui, X.; Eyles, D.W. Vitamin D signaling and the differentiation of developing dopamine systems. Neuroscience 2016, 333, 193–203.
- Cui, X.; Pelekanos, M.; Liu, P.-Y.; Burne, T.H.J.; McGrath, J.J.; Eyles, D.W. The vitamin D receptor in dopamine neurons; its presence in human substantia nigra and its ontogenesis in rat midbrain. Neuroscience 2013, 236, 77–87.
- Veenstra, T.D.; Prüfer, K.; Koenigsberger, C.; Brimijoin, S.W.; Grande, J.P.; Kumar, R. 1,25-Dihydroxyvitamin D3 receptors in the central nervous system of the rat embryo. Brain Res. 1998, 804, 193–205.
- Hawes, J.E.; Tesic, D.; Whitehouse, A.J.; Zosky, G.R.; Smith, J.T.; Wyrwoll, C.S. Maternal vitamin D deficiency alters fetal brain development in the BALB/c mouse. Behav. Brain Res. 2015, 286, 192–200.
- Cui, X.; Pelekanos, M.; Burne, T.H.J.; McGrath, J.J.; Eyles, D.W. Maternal vitamin D deficiency alters the expression of genes involved in dopamine specification in the developing rat mesencephalon. Neurosci. Lett. 2010, 486, 220–223.
- Kesby, J.P.; Cui, X.; Ko, P.; McGrath, J.J.; Burne, T.H.J.; Eyles, D.W. Developmental vitamin D deficiency alters dopamine turnover in neonatal rat forebrain. Neurosci. Lett. 2009, 461, 155–158.
- Cui, X.; Pertile, R.; Liu, P.; Eyles, D.W. Vitamin D regulates tyrosine hydroxylase expression: N-cadherin a possible mediator. Neuroscience 2015, 304, 90–100.
- Bonnin, A.; Levitt, P. Fetal, maternal, and placental sources of serotonin and new implications for developmental programming of the brain. Neuroscience 2011, 197, 1–7.
- Patrick, R.P.; Ames, B.N. Vitamin D hormone regulates serotonin synthesis. Part 1: Relevance for autism. FASEB J. 2014, 28, 2398–2413.
- Patrick, R.P.; Ames, B.N. Vitamin D and the omega-3 fatty acids control serotonin synthesis and action, part 2: Relevance for ADHD, bipolar disorder, schizophrenia, and impulsive behavior. FASEB J. 2015, 29, 2207–2222.
- Kaneko, I.; Sabir, M.S.; Dussik, C.M.; Whitfield, G.K.; Karrys, A.; Hsieh, J.C.; Haussler, M.R.; Meyer, M.B.; Pike, J.W.; Jurutka, P.W. 1,25-DihydroxyVitamin D regulates expression of the tryptophan hydroxylase 2 and leptin genes: Implication for behavioral influences of Vitamin D. FASEB J. 2015, 29, 4023–4035.
- Jiang, P.; Zhang, L.H.; Cai, H.L.; Li, H.D.; Liu, Y.P.; Tang, M.M.; Dang, R.L.; Zhu, W.Y.; Xue, Y.; He, X. Neurochemical effects of chronic administration of calcitriol in rats. Nutrients 2014, 6, 6048–6059.
- Chen, J.; Doroudi, M.; Cheung, J.; Grozier, A.L.; Schwartz, Z.; Boyan, B.D. Plasma membrane Pdia3 and VDR interact to elicit rapid responses to 1α,25(OH)2D3. Cell. Signal. 2013, 25, 2362–2373.
- Landel, V.; Stephan, D.; Cui, X.; Eyles, D.; Feron, F. Differential expression of vitamin D-associated enzymes and receptors in brain cell subtypes. J. Steroid Biochem. Mol. Biol. 2018, 177, 129–134.
- Pendyala, G.; Ninemire, C.; Fox, H.S. Protective role for the disulfide isomerase PDIA3 in methamphetamine neurotoxicity. PLoS ONE 2012, 7, e38909.
- Hetz, C.; Russelakis-Carneiro, M.; Wälchli, S.; Carboni, S.; Vial-Knecht, E.; Maundrell, K.; Castilla, J.; Soto, C. The disulfide isomerase Grp58 is a protective factor against prion neurotoxicity. J. Neurosci. 2005, 25, 2793–2802.
- Yoo, D.Y.; Cho, S.B.; Jung, H.Y.; Kim, W.; Nam, S.M.; Kim, J.W.; Moon, S.M.; Yoon, Y.S.; Kim, D.W.; Choi, S.Y.; et al. Differential roles of exogenous protein disulfide isomerase A3 on proliferating cell and neuroblast numbers in the normal and ischemic gerbils. Brain Behav. 2020, 10, 1–12.
- Wang, W.T.; Sun, L.; Sun, C.H. PDIA3-regulted inflammation and oxidative stress contribute to the traumatic brain injury (TBI) in mice. Biochem. Biophys. Res. Commun. 2019, 518, 657–663.
- Chiavari, M.; Ciotti, G.M.P.; Canonico, F.; Altieri, F.; Lacal, P.M.; Graziani, G.; Navarra, P.; Lisi, L. Pdia3 expression in glioblastoma modulates macrophage/microglia pro-tumor activation. Int. J. Mol. Sci. 2020, 21, 8214.
- Bouillon, R.; Schuit, F.; Antonio, L.; Rastinejad, F. Vitamin D Binding Protein: A Historic Overview. Front. Endocrinol. 2020, 10, 910.
- Bikle, D.D. The Free Hormone Hypothesis: When, Why, and How to Measure the Free Hormone Levels to Assess Vitamin D, Thyroid, Sex Hormone, and Cortisol Status. JBMR Plus 2021, 5, 1–10.
- Marzolo, M.-P.; Farfán, P. New Insights into the Roles of Megalin/LRP2 and the Regulation of its Functional Expression. Biol. Res. 2011, 44, 89–105.
- Xue, Y.; He, X.; Li, H.-D.; Deng, Y.; Yan, M.; Cai, H.-L.; Tang, M.-M.; Dang, R.-L.; Jiang, P. Simultaneous Quantification of 25-Hydroxyvitamin D 3 and 24,25-Dihydroxyvitamin D 3 in Rats Shows Strong Correlations between Serum and Brain Tissue Levels. Int. J. Endocrinol. 2015, 2015, 296531.
- Fu, X.; Dolnikowski, G.G.; Patterson, W.B.; Dawson-Hughes, B.; Zheng, T.; Morris, M.C.; Holland, T.M.; Booth, S.L. Determination of Vitamin D and Its Metabolites in Human Brain Using an Ultra-Pressure LC–Tandem Mass Spectra Method. Curr. Dev. Nutr. 2019, 3, nzz074.
- Stumpf, W.; Sar, M.; Clark, S.; DeLuca, H. Brain target sites for 1,25-dihydroxyvitamin D3. Science 1982, 215, 1403–1405.
- Lee, D.H.; Kang, H.; Kim, J.H.; Jung, M.H.; Cho, M.C. Cerebrospinal fluid vitamin D-binding protein as a new biomarker for the diagnosis of meningitis. Neurol. Sci. 2019, 40, 1597–1605.
- Shah, I.; Akhtar, M.K.; Hisaindee, S.; Rauf, M.A.; Sadig, M.; Ashraf, S.S. Clinical diagnostic tools for vitamin D assessment. J. Steroid Biochem. Mol. Biol. 2018, 180, 105–117.
- Herrmann, M.; Farrell, C.J.L.; Pusceddu, I.; Fabregat-Cabello, N.; Cavalier, E. Assessment of Vitamin D status—A changing landscape. Clin. Chem. Lab. Med. 2017, 55, 3–26.
- Smith, L.M.; Gallagher, J.C. Dietary Vitamin D Intake for the Elderly Population: Update on the Recommended Dietary Allowance for Vitamin D. Endocrinol. Metab. Clin. N. Am. 2017, 46, 871–884.
- Bikle, D.D. Vitamin D Metabolism, Mechanism of Action, and Clinical Applications. Chem. Biol. 2014, 21, 319–329.
- Bikle, D.D.; Schwartz, J. Vitamin D binding protein, total and free Vitamin D levels in different physiological and pathophysiological conditions. Front. Endocrinol. 2019, 10, 317.
- Cordeiro, A.; Ramalho, A. Vitamin D Status in Obesity: Relation with Expression of Vitamin D Receptor and Vitamin D Hydroxylation Enzymes in Subcutaneous and Visceral Adipose Tissue. In Adiposity—Omics and Molecular Understanding; InTech: London, UK, 2017.
- Jenkinson, C. The vitamin D metabolome: An update on analysis and function. Cell Biochem. Funct. 2019, 37, 408–423.
- Saponaro, F.; Saba, A.; Zucchi, R. An update on vitamin d metabolism. Int. J. Mol. Sci. 2020, 21, 6573.
- Bikle, D.D.; Malmstroem, S.; Schwartz, J. Current Controversies. Endocrinol. Metab. Clin. N. Am. 2017, 46, 901–918.
- Wang, Z.; Schuetz, E.G.; Xu, Y.; Thummel, K.E. Interplay between vitamin D and the drug metabolizing enzyme CYP3A4. J. Steroid Biochem. Mol. Biol. 2013, 136, 54–58.
- Bivona, G.; Agnello, L.; Bellia, C.; Iacolino, G.; Scazzone, C.; Lo Sasso, B.; Ciaccio, M. Non-skeletal activities of vitamin d: From physiology to brain pathology. Medicina 2019, 55, 341.
- El-Atifi, M.; Dreyfus, M.; Berger, F.; Wion, D. Expression of CYP2R1 and VDR in human brain pericytes. Neuroreport 2015, 26, 245–248.
- Toselli, F.; Dodd, P.R.; Gillam, E.M.J. Emerging roles for brain drug-metabolizing cytochrome P450 enzymes in neuropsychiatric conditions and responses to drugs. Drug Metab. Rev. 2016, 48, 379–404.
- Kuban, W.; Daniel, W.A. Cytochrome P450 expression and regulation in the brain. Drug Metab. Rev. 2021, 53, 1–29.
- Christakos, S.; Dhawan, P.; Verstuyf, A.; Verlinden, L.; Carmeliet, G. Vitamin D: Metabolism, molecular mechanism of action, and pleiotropic effects. Physiol. Rev. 2015, 96, 365–408.
- Bikle, D.D.; Patzek, S.; Wang, Y. Physiologic and pathophysiologic roles of extra renal CYP27b1: Case report and review. Bone Rep. 2018, 8, 255–267.
- Eyles, D.W. Vitamin D: Brain and Behavior. JBMR Plus 2021, 5, e10419.
- Reddy, G.S.; Tserng, K.Y. Calcitroic acid, end product of renal metabolism of 1,25-dihydroxyvitamin D3 through the C-24 oxidation pathway. Biochemistry 1989, 28, 1763–1769.
- Veldurthy, V.; Wei, R.; Campbell, M.; Lupicki, K.; Dhawan, P.; Christakos, S. 25-Hydroxyvitamin D3 24-Hydroxylase: A key regulator of 1, 25 (OH) 2D3 catabolism and calcium homeostasis. Vitam. Horm. 2016, 100, 137–150.
- Slominski, A.T.; Kim, T.K.; Li, W.; Postlethwaite, A.; Tieu, E.W.; Tang, E.K.Y.; Tuckey, R.C. Detection of novel CYP11A1-derived secosteroids in the human epidermis and serum and pig adrenal gland. Sci. Rep. 2015, 5, 14875.
- Yu, O.B.; Arnold, L.A. Calcitroic Acid-A Review. ACS Chem. Biol. 2016, 11, 2665–2672.
- Ishizuka, S.; Kurihara, N.; Hiruma, Y.; Miura, D.; Namekawa, J.; Tamura, A.; Kato-Nakamura, Y.; Nakano, Y.; Takenouchi, K.; Hashimoto, Y.; et al. 1α,25-Dihydroxyvitamin D3-26,23-lactam analogues function as vitamin D receptor antagonists in human and rodent cells. J. Steroid Biochem. Mol. Biol. 2008, 110, 269–277.
- Anderson, P.H. Vitamin D Activity and Metabolism in Bone. Curr. Osteoporos. Rep. 2017, 15, 443–449.
- Smolders, J.; Schuurman, K.G.; Van Strien, M.E.; Melief, J.; Hendrickx, D.; Hol, E.M.; Van Eden, C.; Luchetti, S.; Huitinga, I. Expression of vitamin D receptor and metabolizing enzymes in multiple sclerosis-affected brain tissue. J. Neuropathol. Exp. Neurol. 2013, 72, 91–105.
- Burne, T.H.J.; Becker, A.; Brown, J.; Eyles, D.W.; Mackay-Sim, A.; McGrath, J.J. Transient prenatal Vitamin D deficiency is associated with hyperlocomotion in adult rats. Behav. Brain Res. 2004, 154, 549–555.
- Kesby, J.P.; Burne, T.H.J.; McGrath, J.J.; Eyles, D.W. Developmental Vitamin D Deficiency Alters MK 801-Induced Hyperlocomotion in the Adult Rat: An Animal Model of Schizophrenia. Biol. Psychiatry 2006, 60, 591–596.
- Becker, A.; Eyles, D.W.; McGrath, J.J.; Grecksch, G. Transient prenatal vitamin D deficiency is associated with subtle alterations in learning and memory functions in adult rats. Behav. Brain Res. 2005, 161, 306–312.
- Grecksch, G.; Rüthrich, H.; Höllt, V.; Becker, A. Transient prenatal vitamin D deficiency is associated with changes of synaptic plasticity in the dentate gyrus in adult rats. Psychoneuroendocrinology 2009, 34, 258–264.
- Harms, L.R.; Eyles, D.W.; McGrath, J.J.; Mackay-Sim, A.; Burne, T.H.J. Developmental vitamin D deficiency alters adult behaviour in 129/SvJ and C57BL/6J mice. Behav. Brain Res. 2008, 187, 343–350.
- Harms, L.R.; Turner, K.M.; Eyles, D.W.; Young, J.W.; McGrath, J.J.; Burne, T.H.J. Attentional processing in C57BL/6J mice exposed to developmental vitamin D deficiency. PLoS ONE 2012, 7, 3–12.
- de Abreu, D.A.F.; Nivet, E.; Baril, N.; Khrestchatisky, M.; Roman, F.; Féron, F. Developmental vitamin D deficiency alters learning in C57Bl/6J mice. Behav. Brain Res. 2010, 208, 603–608.
- Turner, K.M.; Young, J.W.; McGrath, J.J.; Eyles, D.W.; Burne, T.H.J. Cognitive performance and response inhibition in developmentally vitamin D (DVD)-deficient rats. Behav. Brain Res. 2013, 242, 47–53.
- Overeem, K.; Alexander, S.; Burne, T.H.J.; Ko, P.; Eyles, D.W. Developmental vitamin D deficiency in the rat impairs recognition memory, but has no effect on social approach or hedonia. Nutrients 2019, 11, 2713.
- Antunes, M.; Biala, G. The novel object recognition memory: Neurobiology, test procedure, and its modifications. Cogn. Process. 2012, 13, 93–110.
- Li, Y.C.; Pirro, A.E.; Amling, M.; Delling, G.; Baron, R.; Bronson, R.; Demay, M.B. Targeted ablation of the vitamin D receptor: An animal model of vitamin D-dependent rickets type II with alopecia. Proc. Natl. Acad. Sci. USA 1997, 94, 9831–9835.
- Yoshizawa, T.; Handa, Y.; Uematsu, Y.; Takeda, S.; Sekine, K.; Yoshihara, Y.; Kawakami, T.; Arioka, K.; Sato, H.; Uchiyama, Y.; et al. Mice lacking the vitamin D receptor exhibit impaired bone formation, uterine hypoplasia and growth retardation after weaning. Nat. Genet. 1997, 16, 391–396.
- Van Cromphaut, S.J.; Dewerchin, M.; Hoenderop, J.G.J.; Stockmans, I.; Van Herck, E.; Kato, S.; Bindels, R.J.M.; Collen, D.; Carmeliet, P.; Bouillon, R.; et al. Duodenal calcium absorption in vitamin D receptor-knockout mice: Functional and molecular aspects. Proc. Natl. Acad. Sci. USA 2001, 98, 13324–13329.
- Kaufmann, M.; Lee, S.M.; Pike, J.W.; Jones, G. VDR-Expressing Transgenes Normalize Serum Vitamin Cyp24a1 Expression in VDR Null Mice. Endocrinology 2016, 156, 4388–4397.
- Li, Y.C.; Amling, M.; Pirro, A.E.; Priemel, M.; Meuse, J.; Baron, R.; Delling, G.; Demay, M.B. Normalization of mineral ion homeostasis by dietary means prevents hyperparathyroidism, rickets, and osteomalacia, but not alopecia in vitamin D receptor-ablated mice. Endocrinology 1998, 139, 4391–4396.
- Kollenkirchen, U.; Fox, J.; Walters, M.R. Normocalcemia without hyperparathyroidism in vitamin D-deficient rats. J. Bone Miner. Res. 1991, 6, 273–278.
- Song, Y.; Kato, S.; Fleet, J.C. Vitamin D Receptor (VDR) Knockout Mice Reveal VDR-Independent Regulation of Intestinal Calcium Absorption and ECaC2 and Calbindin D9k mRNA. J. Nutr. 2003, 133, 374–380.
- Shimada, T.; Yamazaki, Y.; Takahashi, M.; Hasegawa, H.; Urakawa, I.; Oshima, T.; Ono, K.; Kakitani, M.; Tomizuka, K.; Fujita, T.; et al. Vitamin D receptor-independent FGF23 actions in regulating phosphate and vitamin D metabolism. Am. J. Physiol. Physiol. 2005, 289, F1088–F1095.
- David, V.; Dai, B.; Martin, A.; Huang, J.; Han, X.; Quarles, L.D. Calcium Regulates FGF-23 Expression in Bone. Endocrinology 2013, 154, 4469–4482.
- Liu, P.; Chen, L.; Bai, X.; Karaplis, A.; Miao, D.; Gu, N. Impairment of spatial learning and memory in transgenic mice overexpressing human fibroblast growth factor-23. Brain Res. 2011, 1412, 9–17.
- Grundmann, S.M.; Brandsch, C.; Rottstädt, D.; Kühne, H.; Stangl, G.I. The high calcium, high phosphorus rescue diet is not suitable to prevent secondary hyperparathyroidism in vitamin D receptor deficient mice. Front. Physiol. 2017, 8, 212.
- Burne, T.H.J.; Johnston, A.N.B.; McGrath, J.J.; MacKay-Sim, A. Swimming behaviour and post-swimming activity in Vitamin D receptor knockout mice. Brain Res. Bull. 2006, 69, 74–78.
- Kalueff, A.V.; Keisala, T.; Minasyan, A.; Kuuslahti, M.; Miettinen, S.; Tuohimaa, P. Behavioural anomalies in mice evoked by “Tokyo” disruption of the Vitamin D receptor gene. Neurosci. Res. 2006, 54, 254–260.
- Keisala, T.; Minasyan, A.; Järvelin, U.; Wang, J.; Hämäläinen, T.; Kalueff, A.V.; Tuohimaa, P. Aberrant nest building and prolactin secretion in vitamin D receptor mutant mice. J. Steroid Biochem. Mol. Biol. 2007, 104, 269–273.
- Kalueff, A.V.; Lou, Y.-R.; Laaksi, I.; Tuohimaa, P. Increased anxiety in mice lacking vitamin D receptor gene. Neuroreport 2004, 15, 1271–1274.
- Tishkoff, D.X.; Nibbelink, K.A.; Holmberg, K.H.; Dandu, L.; Simpson, R.U. Functional Vitamin D Receptor (VDR) in the T-Tubules of Cardiac Myocytes: VDR Knockout Cardiomyocyte Contractility. Endocrinology 2008, 149, 558–564.
- Izzo, M.; Carrizzo, A.; Izzo, C.; Cappello, E.; Cecere, D.; Ciccarelli, M.; Iannece, P.; Damato, A.; Vecchione, C.; Pompeo, F. Vitamin D: Not just bone metabolism but a key player in cardiovascular diseases. Life 2021, 11, 452.
- Wong, K.E.; Szeto, F.L.; Zhang, W.; Ye, H.; Kong, J.; Zhang, Z.; Sun, X.J.; Li, Y.C. Involvement of the vitamin D receptor in energy metabolism: Regulation of uncoupling proteins. Am. J. Physiol. Metab. 2009, 296, E820–E828.
- Byrne, J.H.; Voogt, M.; Turner, K.M.; Eyles, D.W.; McGrath, J.J.; Burne, T.H.J. The Impact of Adult Vitamin D Deficiency on Behaviour and Brain Function in Male Sprague-Dawley Rats. PLoS ONE 2013, 8, e71593.
- Keeney, J.T.R.; Förster, S.; Sultana, R.; Brewer, L.D.; Latimer, C.S.; Cai, J.; Klein, J.B.; Porter, N.M.; Butterfield, D.A. Dietary vitamin D deficiency in rats from middle to old age leads to elevated tyrosine nitration and proteomics changes in levels of key proteins in brain: Implications for low vitamin D-dependent age-related cognitive decline. Free Radic. Biol. Med. 2013, 65, 324–334.
- Brouwer-Brolsma, E.M.; Schuurman, T.; de Groot, L.C.P.M.G.; Feskens, E.J.M.; Lute, C.; Naninck, E.F.G.; Arndt, S.S.; van der Staay, F.J.; Bravenboer, N.; Korosi, A.; et al. No role for vitamin D or a moderate fat diet in aging induced cognitive decline and emotional reactivity in C57BL/6 mice. Behav. Brain Res. 2014, 267, 133–143.
- Latimer, C.S.; Brewer, L.D.; Searcy, J.L.; Chen, K.C.; Popović, J.; Kraner, S.D.; Thibault, O.; Blalock, E.M.; Landfield, P.W.; Porter, N.M. Vitamin D prevents cognitive decline and enhances hippocampal synaptic function in aging rats. Proc. Natl. Acad. Sci. USA 2014, 111, E4359–E4366.
- Liang, Q.; Cai, C.; Duan, D.; Hu, X.; Hua, W.; Jiang, P.; Zhang, L.; Xu, J.; Gao, Z. Postnatal Vitamin D Intake Modulates Hippocampal Learning and Memory in Adult Mice. Front. Neurosci. 2018, 12, 141.
More