Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Marc S. Halfon and Version 2 by Peter Tang.
The REDflyREDfly (Regulatory Element Database for Fly) (Regulatory Element Database for Fly) database (http://redfly.ccr.buffalo.edu/, accessed on 27 July 2022) integrates all of the available insect cis-regulatory information from multiple sources to provide a comprehensive collection of known regulatory elements.
- insects
- Drosophila
- regulatory genomics
- gene regulation
- cis-regulatory module
- enhancer
- genome annotation
1. Introduction
The turn-of-the-century advent of fully sequenced metazoan genomes brought with it the first genome annotations, which were largely confined to positions of confirmed and predicted genes, and typically housed in community-specific model-organism databases, e.g., [1][2][3][4][1,2,3,4]. Remarkably, over two decades later, the major databases (see [5]) are still mostly lacking annotation of non-coding regulatory sequences. These sequences include distal “cis-regulatory modules” (CRMs), a generic term encompassing such regulatory elements as enhancers, which mediate positive gene regulation; silencers, involved in negative regulation; and a growing number of additional elements that are not easily classified including PREs, super-enhancers, insulators, tethering elements, and others [6][7][8][9][10][11][6,7,8,9,10,11].
2. Utility of REDfly
Prior to the development of REDfly, large-scale analyses of regulatory sequences were challenging to conduct, as the bulk of the existing regulatory data was distributed among hundreds of individual publications. Consequently, what few analyses were completed were performed on small and frequently biased sets of CRMs, such as a limited subset of early developmental pair-rule stripe enhancers in Drosophila, e.g., [12][13][14][16,17,18]. By curating these data and making them findable, accessible, interoperable, and usable (FAIR) [15][19], REDfly made it possible to bring statistical, computational, and comparative genomics methods to bear on their study. REDfly enabled the first-ever large-scale, relatively unbiased analysis of CRMs, which immediately revealed novel insights into CRM-sequence composition, differences among tissue-specific groups of CRMs, and an early indication of the presence of enhancer RNAs (eRNAs) as a prevalent CRM characteristic [16][20]. REDfly, by continuing to compile the data from hundreds and eventually thousands of individual experiments scattered throughout four decades of literature, subsequently proved instrumental in facilitating studies in a wide variety of research areas, including: Biology of CRMs. REDfly has been used to investigate the organization of TFBSs within CRMs [17][21] and how combinatorial binding influences CRM activity [18][22]. Soluri et al. [19][23] investigated how pioneer TFs control chromatin accessibility, and Blick et al. [20][24] examined the ability of CRMs to act in trans. REDfly data helped to illustrate how CRMs can have multiple functions [21][25], such as dual use as both enhancers and Polycomb response elements [22][26], or as both enhancers and silencers [23][27]. Interpretation of genomic data. REDfly has been critical for interpreting data from large-scale genomics projects including TF binding studies, e.g., [24][25][28,29] and studies of insulators [26][27][30,31]. A study challenging the understanding of which epigenetic marks characterize regulatory sequences depended on REDfly data [28][32]. REDfly has been used to study chromosome domains and chromatin “states”, e.g., [29][30][31][32][33,34,35,36], to explore 3D-chromatin conformation [33][34][37,38], to study ncRNA and eRNA expression [35][36][39,40], and to validate scATAC-seq approaches, e.g., [37][38][41,42]. Computational CRM discovery. REDfly has played a dramatic role in methods for computational CRM discovery, both as a source of training data and as a method for validating predictions, e.g., [39][40][41][42][43][44][45][46][47][43,44,45,46,47,48,49,50,51]. Su et al. [48][52] used REDfly data to assess CRM-discovery approaches, which would have been impossible without REDfly. Computational CRM-discovery methods using REDfly for training data also can identify CRMs in diverse insect species [49][53] and, as such, provide a powerful tool for annotating insect regulatory genomes [50][54]. CRM evolution. REDfly has enabled studies of CRM evolution and TFBS turnover, e.g., [51][52][53][54][55][56][57][55,56,57,58,59,60,61]. Wang et al. [58][62] used REDfly data to investigate the selective pressure on DNA shape at TF binding sites, and Peng et al. [59][63] explored the relationship between chromatin accessibility and TF binding to predict evolutionary changes in enhancer activity. As can be seen from these examples, REDfly is an important source of raw data for analysis, hypothesis generation, assessment, validation, and empirical research.3. REDfly Data Model
REDfly curates two types of data: CRMs and transcription factor binding sites (TFBSs), with CRMs being the main focus. Historically, CRM annotations have been drawn from reporter gene assays in transgenic animals or cultured cells, but an increasingly diverse set of assays are starting to be included. In particular, several years ago REDfly started to capture CRMs identified through various “X-seq” assays, such as ATAC-seq, FAIRE-seq, DNase-seq, ChIP-seq, etc., as well as from purely computational predictions, in recognition of the fact that many regulatory sequences are presently being defined by these methods. There is considerable debate in the regulatory genomics field as to just how CRMs should be defined, with several studies indicating that the different methods of CRM identification have led to widely non-overlapping results and raising questions as to which, if any, of these methods most accurately identify CRMs [60][61][62][64,65,66]. As a result, REDfly separates its CRM data into four distinct subclasses: reporter constructs (RC), CRM_segments, predicted CRMs (pCRM), and inferred CRMs (iCRM). RCs and CRM_segments are drawn from activity-based assays (Figure 1, left), primarily gene-expression data from either reporter genes or from native genes following mutation or deletion of regulatory sequences. RCs mainly represent reporter-gene results, assayed either in transgenic animals or in cell-culture assays; the two types of assays can be independently searched. The CRM_segment class contains sequences that are demonstrated to be necessary for gene regulation but, unlike in a reporter gene assay, are not necessarily sufficient. Such sequences can be obtained from the analysis of small chromosomal deletions or site-directed mutagenesis but are increasingly being found in the literature as a result of CRISPR/Cas9-mediated targeted sequence deletions. pCRMs, on the other hand, reflect CRMs identified by assays that do not require demonstration of activity, for example, the presence of histone modifications, or computational predictions (Figure 1, right). iCRMs are not curated from the literature, but represent putative regulatory elements based on the analysis of other REDfly data (see below).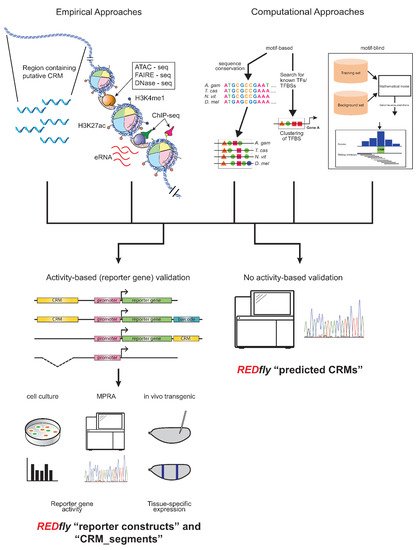
Figure 1. Activity-based and non-activity-based methods for defining regulatory sequences. Top: A wide variety of methods exist for identifying regulatory sequences based on both empirical (left) and computational (right) approaches. Empirical approaches include unbiased testing of non-coding DNA regions as well as selection of sequences based on chromatin accessibility, histone post-translational modification, transcription factor binding, production of enhancer RNAs, and others. Computational approaches may include assessment of sequence conservation, presence of transcription factor binding motifs, or various machine-learning methods. Bottom: Results from these regulatory element discovery methods can be obtained with (left) or without (right) the use of activity-based criteria. Activity-based criteria typically involve some sort of reporter-gene assay, which might be performed in cultured cells, using next-generation sequencing in a “massively parallel reporter assay” (MPRA), or in transgenic animals; recently, testing via genomic deletion via CRISPR/Cas9 has also been gaining popularity. REDfly classifies regulatory sequences derived from these methods as reporter constructs (RCs) and CRM_segments, while any methods that identify regulatory sequences without recourse to activity-based criteria are referred to as predicted CRMs (pCRMs). These somewhat historical definitions should not be construed to imply that one or the other type of data is more accurate or “correct”.
4. Species in REDfly
Although REDfly has historically been focused on Drosophila melanogaster, the clear value of comparative genomics and of working with non-traditional model organisms, a vast increase in the number of sequenced insect genomes, and the small but growing availability of both predicted and validated insect regulatory sequences has led us to expand REDfly by implementing the ability to curate regulatory sequences for additional insect species. Information on which species are represented can be found on the “Species” page; current species include the mosquitoes Anopheles gambiae and Aedes aegypti and the beetle Tribolium castaneum. Additional species will be added as data accumulate. Since insect CRMs are often tested using transgenic Drosophila, REDfly divides the sequence and gene-feature data and the expression pattern and cell-line data into separate components. Each REDfly record has both a “sequence from” and an “assayed in” component. Sequence and gene-feature data are linked to the former, and anatomy and staging data to the latter. While it is preferable to describe species-specific reporter-gene-expression patterns using the proper species-specific anatomy ontology, many species lack an ontology as rich in terms as that for Drosophila. Therefore, terms from the Drosophila ontology can also be used to annotate expression in other species.
In order to facilitate research using these newly added genomes, the researchers have implemented interfaces for BLAT [63][70] and in silico PCR [64][71] for each species included in REDfly. These can be accessed through the “Species” page.
5. Contents of REDfly
REDfly has continued to expand its contents at a rapid rate (Table 1). Since the end of 2019, the number of curated publications has increased by 30%, leading to an increase in the total number of Reporter Construct records by over 25% and in the number of pCRMs by almost 60%. Not reflected in these numbers, however, is an ambitious endeavor to update all RC records with the full set of RC expression attributes (developmental staging, sexually dimorphic activity, ectopic activity, and enhancer/silencer activity), which did not become a full part of the REDfly data model until the release of REDfly v6 in 2020. Since that time, over one-third of the RC records have been updated to contain this information.
Table 1.
REDfly contents as of 1 July 2022.
|
Reporter Constructs (RCs) |
43,819 |
|
From in vivo reporter genes |
21,690 |
|
Associated with staging and other attributes |
17,961 |
|
CRM_segments |
16 |
|
Predicted CRMs (pCRMs) |
14,318 |
|
Inferred CRMs (iCRMs) |
7760 |
|
Transcription Factor Binding Sites (TFBS) |
2717 |
|
Publications curated |
1366 |
6. Using REDfly
The extensive data and metadata REDfly provides for each of its records allows for detailed customized searching of the database contents. Typical entry points for a REDfly search are via a gene name (Figure 2A(a)) or a literature reference (via PubMed ID, Figure 2A(b)). By default, searching for a gene name will execute a “by locus” search in which any elements annotated as being associated with that gene, as well as any elements found within a user-customizable range of 10 kb upstream or downstream of the gene (regardless of assigned target gene), will be returned. Moreover, by default, elements identified solely by assays performed in cultured cells are omitted from the results (Figure 2A(d)); unchecking the check-box causes these results to be included.
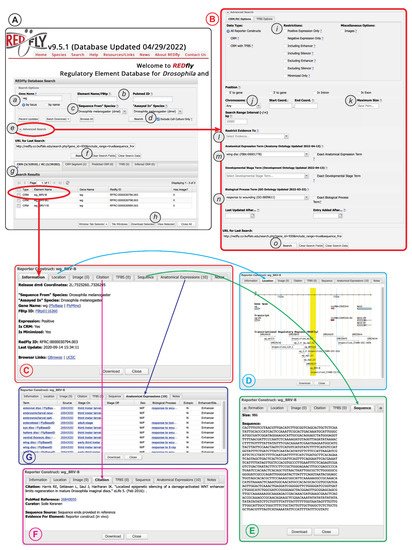
Figure 2. The REDfly search interface. See text for details. (A) The basic search panel. (B) The Advanced Search panel. (C) The Detailed Results “Information” pane. (D) The Detailed Results “Location” pane. (E) The Detailed Results “Sequence” pane. (F) The Detailed Results “Citation” pane. (G) The Detailed Results “Anatomical Expression” pane.
Clicking on the “Advanced Search” arrow (Figure 2A(e)) allows access to a large variety of additional options (Figure 2B), including the ability to restrict searches to specific RC attributes, genomic locations, anatomical regions, developmental stages, or biological processes. More detailed and complex search capabilities are under development.
Regardless of whether “basic” or “advanced” search is used, a summary of the results will appear in the “Search Results” pane directly below the main search window (Figure 2A(g)). Results for each REDfly data class—RCs/CRMs, CRM_segments, pCRMs, TFBSs, and iCRMs—are displayed in individual tabs to make it easier to view results by type. Checkboxes allow selection of records for download (Figure 2A(h)) in any of a number of convenient formats. Alternatively, clicking on an individual result will open a multipaned “Detailed View” window containing full information for the selected record (Figure 2C). Basic location and attribute data are displayed in the “Information” tab, along with links to relevant model-organism databases and genome browsers (Figure 2C). The “Location” tab provides a snapshot of the element in its genomic milieu (Figure 2D). The “Sequence” tab displays the genomic sequence and its size (Figure 2E), while the “Citation” tab (Figure 2F) provides a citation and link to the publication describing the current element, plus a description of the evidence used by REDfly curators to annotate sequence and expression information. The “Anatomical Expression” tab (Figure 2G) lists each cell type or tissue where the regulatory element is active, along with a specific citation for that activity data (since activity data may be drawn from multiple references), developmental staging for the observed activity, and the other attributes discussed above, e.g., sexually dimorphic activity, ectopic expression, and enhancer or silencer activity.
Since CRM activity can be complex and not easily summarized using the anatomical and staging terms available in the relevant ontologies, the researchers also supply a “Notes” tab containing details and clarifications.