Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Lindsay Dong and Version 1 by Rebecca C. Deed.
Volatile polyfunctional thiol compounds, particularly 3-sulfanylhexan-1-ol (3SH) and 3-sulfanylhexyl acetate (3SHA), are key odorants contributing to the aroma profile of many wine styles, generally imparting tropical grapefruit and passionfruit aromas. 3SH and 3SHA are present in negligible concentrations in the grape berry, juice, and must, suggesting that they are released from non-volatile precursors present in the grape. The exploration of the nature and biogenesis of these precursors to 3SH and 3SHA has proven important for the elucidation of polyfunctional thiol biogenesis during alcoholic fermentation.
- aroma precursors
- calibration methods
- grape wines
- liquid chromatography–mass spectrometry
1. Introduction
Wine aroma is the result of complex interactions between the components of the wine matrix. Although a large proportion of volatile aroma compounds are present only in trace amounts (e.g., ng L−1), many of these compounds have a high sensory impact and further influence the perception of each other by enhancing, suppressing, or adding to the overall aroma profile. To identify and quantify these trace aroma compounds, they first need to be separated from the complex wine matrix. This requirement has led to the uptake of chromatographic systems coupled to various detectors for their subsequent analysis. In the quest to characterize wine aroma compounds, the most appropriate detector, in the first instance, is olfaction. Volatile polyfunctional thiol compounds, including 3-sulfanylhexan-1-ol (3SH), 3-sulfanylhexylacetate (3SHA), and 4-methyl-4-sulfanylpentan-2-one (4MSP) (Figure 1), were first identified in wines using a gas chromatography–olfactometry (GC-O) instrument [1,2]. The comparison of retention times and fragmentation patterns using gas chromatography–mass spectrometry (GC-MS) allowed the identification of the compounds involved in the aroma zones identified by GC-O [1,2], and the investigation into the role of polyfunctional thiols in wine aroma began.
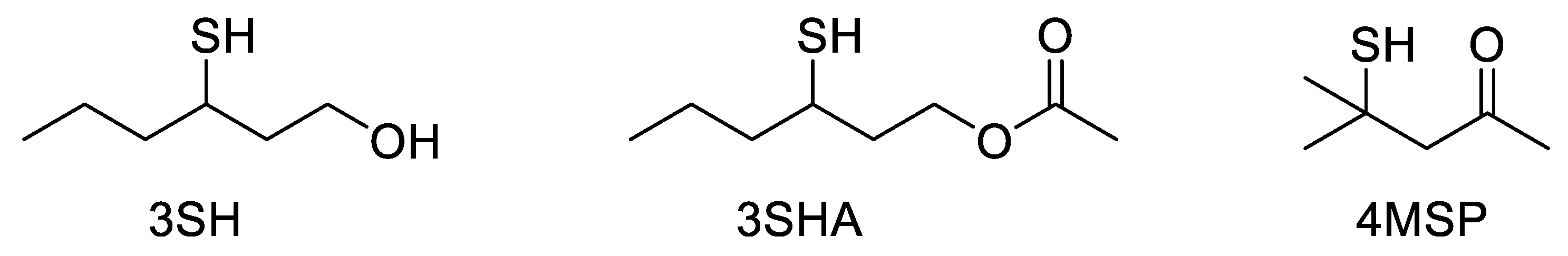
Figure 1. The three key volatile thiols in wine: 3-sulfanylhexan-1-ol (3SH), 3-sulfanylhexylacetate (3SHA), and 4-methyl-4-sulfanylpentan-2-one (4MSP).
2. Analytical Methods to Analyze Wine Aroma Precursors
2.1. Early Exploration of 3SH Precursors: Choosing an Analytical Instrument
The first methods used for the exploration of potential precursors to 3SH and 3SHA used GC-MS instruments. Even before the first formal identification of 3SH in Sauvignon blanc wine [1], it was noted that volatile thiols were not present in the grape must, but that the fermentation of a model medium supplemented with Sauvignon blanc must extracts was able to release 4MSP, a closely related compound [24]. The first precursors to 3SH to be identified were the cysteine conjugates to the thiol (both diastereomers) (Figure 2), which had previously been proposed as precursors to similar sulfur-containing compounds in other plants, such as cabbage and onions [25,26]. Given that cysteine is a biological source of sulfur [27], it was hypothesized that this could provide the thiol moiety of these volatile thiols. As there are a number of cysteine sulfoxide conjugates that can release thiols when metabolized, these also needed to be investigated as potential precursors [22,25,28]. The cysteine S-conjugates (C3SH and cysteine sulfoxide S-conjugates) of the target thiols were identified as one group of precursors to 3SH and 3SHA by comparison of the products of synthetic compounds and grape extracts after incubation with a cysteine β-lyase or alliin lyase, an enzyme specific for the cleavage of cysteine sulfoxide S-conjugates (Scheme 1) [22].
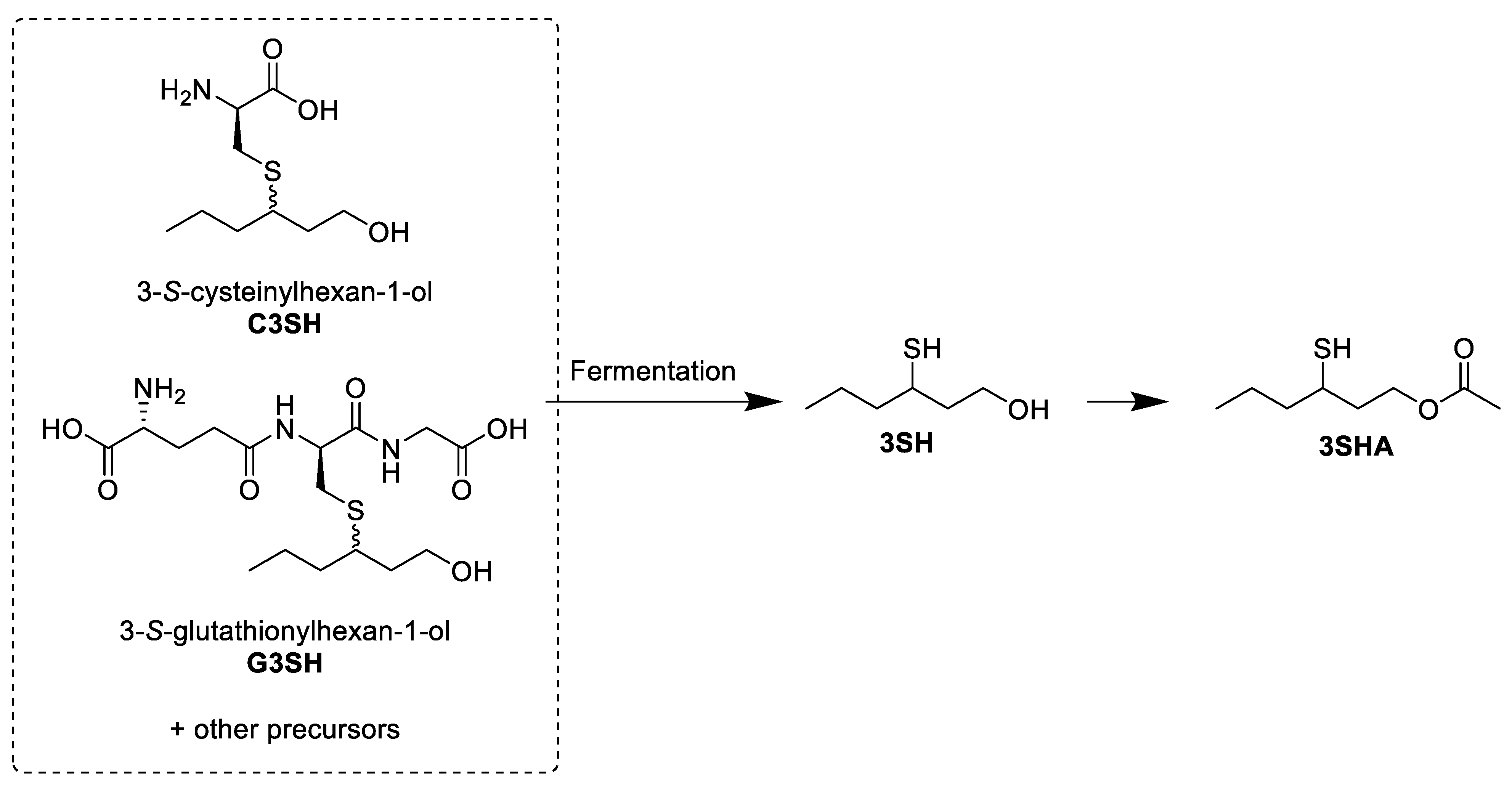
Scheme 1. Simplified visual representation of the work of Tominaga et al. [22], elucidating the nature of the cysteine conjugate precursor by an enzyme assay.
2.2. Gas Chromatography–Mass Spectrometry in 3SH Precursor Analysis
These early explorations of 3SH biogenesis involved both direct and indirect analysis of C3SH, or, more accurately, precursors of 3SH, as it had not been shown that C3SH was the only precursor measured using indirect methods. Indirect strategies depended on the enzymatic release of the volatile thiols from must extracts using established methods to measure the original thiol precursor concentrations [22,29,30,31]. The use of enzymes brought with it several challenges, not least the variation in enzyme activity leading to variability between batches [29]. To mitigate enzyme variability, an internal standard (IS) was used to give an indication of enzyme activity and allow the conversion of the thiol precursors to be compared with the conversion of the IS. However, it was suggested, in early work, that accurate quantification could not be obtained without stable isotopically labeled IS compounds due to the complexity of the matrix and the dangers of assuming that similar compounds will behave in the same way during sample preparation and analysis [32]. The need to utilize a stable-isotope-labeled IS also dictated the requirement to use a mass spectrometer as the detector. Direct approaches mitigating the variability in sample preparation were also developed, improving the detection of the cysteinylated thiol precursors by GC-MS. To analyze molecules via GC-MS, the analytes must be volatile at the raised temperatures used in GC analysis, (around 200 to 350 °C) and be thermally stable [33]. To analyze non-volatile precursors of 3SH, the use of GC-MS required chemical derivatization to volatilize the target analyte. Unlike the indirect approaches that were initially explored, the derivatization of the cysteinylated precursors permitted the direct detection of these compounds, albeit with labor-intensive sample preparation [34]. The typical procedure for the direct analysis of C3SH using derivatization and GC-MS required only 500 µL of sample from which the cysteinylated precursors were isolated on an immobilized copper column before derivatization [22,34,35]. The derivatization step was optimized, and both silylation and perfluoro-acylation were evaluated before heptafluro-acylation (HFA) was ultimately selected as the derivatization method of choice. This method was chosen, as the HF derivatives of the C3SH diastereomers could be chromatographically separated [35]. The determination of the diastereomeric ratio of C3SH is of particular interest, since the R and S enantiomers of 3SH have different perception thresholds (50 ng L−1 and 60 ng L−1) and differ slightly in their aroma descriptors [36].
2.3. Initial 3SH Precursor Analysis with Liquid Chromatography–Mass Spectrometry
It was not until 2008 that LC systems coupled with MS detectors began to be utilized for the analysis of C3SH and other thiol precursors [37], but, from then, they became the standard method. The main advantage of LC-MS for thiol precursor analysis was the reduction in sample preparation required before analysis. The sensitivity of the methods can be compared by looking at the parameters reported for the method validation. The limit of detection (LOD) and limit of quantification (LOQ) are commonly reported measures of method sensitivity based on the signal (S)-to-noise (N) ratio of the method. The definition typically used by the wine research community is that the LOD is the concentration at which the analyte can be reliably detected in the sample (S/N = 3), and the LOQ is the concentration at which the analyte can be reliably quantified in the sample (S/N = 10).3. Development of LC-MS Methods for 3SH Precursor Analysis
3.1. Sample Preparation
Unlike GC-MS systems, LC-MS systems can analyze complex aqueous matrices, such as grape juice and wine, sometimes without sample treatment or purification before injection. Having said this, it can be advantageous to conduct some sample preparation to reduce noise and improve the LOD and LOQ of the analytes [47], in addition to protecting the system from exposure to matrix components, which may compromise the analysis. Many different sample preparation methods have been utilized throughout the literature. Some are as simple as filtering the sample prior to injection, to avoid damaging the HPLC column [42,43,45], while others involve more extensive purification of the target analytes from the rest of the matrix [37,40,41,42,44]. Unsurprisingly, sample preparation methods are significantly influenced by the sample matrix. Even within the field of thiol biogenesis in wine, there are a range of matrices to be studied, from the grape berries and other solid plant matter to the grape juice, must, and wine. Each poses its own challenges, with its own major components of the matrix to remove before analysis. A summary of the sample matrix and preparation methods from previous studies analyzing thiol precursors is shown in Table 2. When analyzing thiol precursors in solid samples, for example, grape skins, seeds, leaves, or commercially available tannins, the precursors need to be extracted into a liquid phase before analysis [46,48,49]. When developing methods involving extraction from solid matrices, the major challenge is optimizing the extraction solvent and conditions to maximize the extraction of the target analytes, standardizing the procedure so that these analytes can be quantified [49]. For thiol precursors in liquid matrices, such as grape juice or wine, further purification is typically required before analysis. The high concentrations of non-target compounds, such as glucose and fructose in grape juice and sweet wine styles, pose both technical and analytical challenges [47]. Such compounds can be insoluble in the mobile phases, may damage the LC column, and can lead to deposits in key components of the system, particularly the MS source. Further, their variation in composition between each sample can lead to significant matrix effects, where the co-elution of non-volatile or non-target compounds leads to the suppression or enhancement of ionization, reducing the reliability of the method and worsening the sensitivity [47]. Sample preparation can strongly influence the reliability of the method and should be standardized within a given method to ensure consistency in the extraction of the target analytes. However, the data available in the literature for analytical methods targeting thiol precursors do not suggest that there is a significant advantage in extensive sample preparation. Although solid-phase extraction (SPE) is a simple method for extracting the target analytes from many matrix components that may interfere with analysis, effectively simplifying and standardizing the sample matrix, it does not offer a significant improvement in method sensitivity. One reason for this is that the solid phase used during SPE for the analysis of these thiol precursors is often substantially similar to the solid phase of the LC column, which would mean that the compounds extracted along with the target analytes are those that will behave similarly in the LC system and are thus likely to co-elute and interfere with the signal [50].3.2. Choice of Calibration Method
To accurately measure the concentrations of the target compounds, a calibration must be conducted. The earliest attempt to quantify volatile thiol concentrations in wines utilized GC-O and involved sequential dilution until the perception threshold of the compound was reached [32]. However, this technique is not feasible for quantifying the non-volatile precursors to these compounds. Even then, the authors noted that the reeliable quantitation of compounds would best be achieved using a MS detector and a stable-isotope-labeled analogue of the target compound [32]. Unfortunately, stable-isotope-labeled compounds tend to be expensive and are often commercially unavailable, meaning that researchers must synthesize them, adding both complexity and cost to the process. Since these early studies, four main quantification techniques, i.e., external calibration (EC), standard addition (SA), internal standard calibration (ISC), and the stable isotope dilution assay (SIDA), have been used to quantify 3SH precursors.4. Further Exploration of the Pathways to 3SH
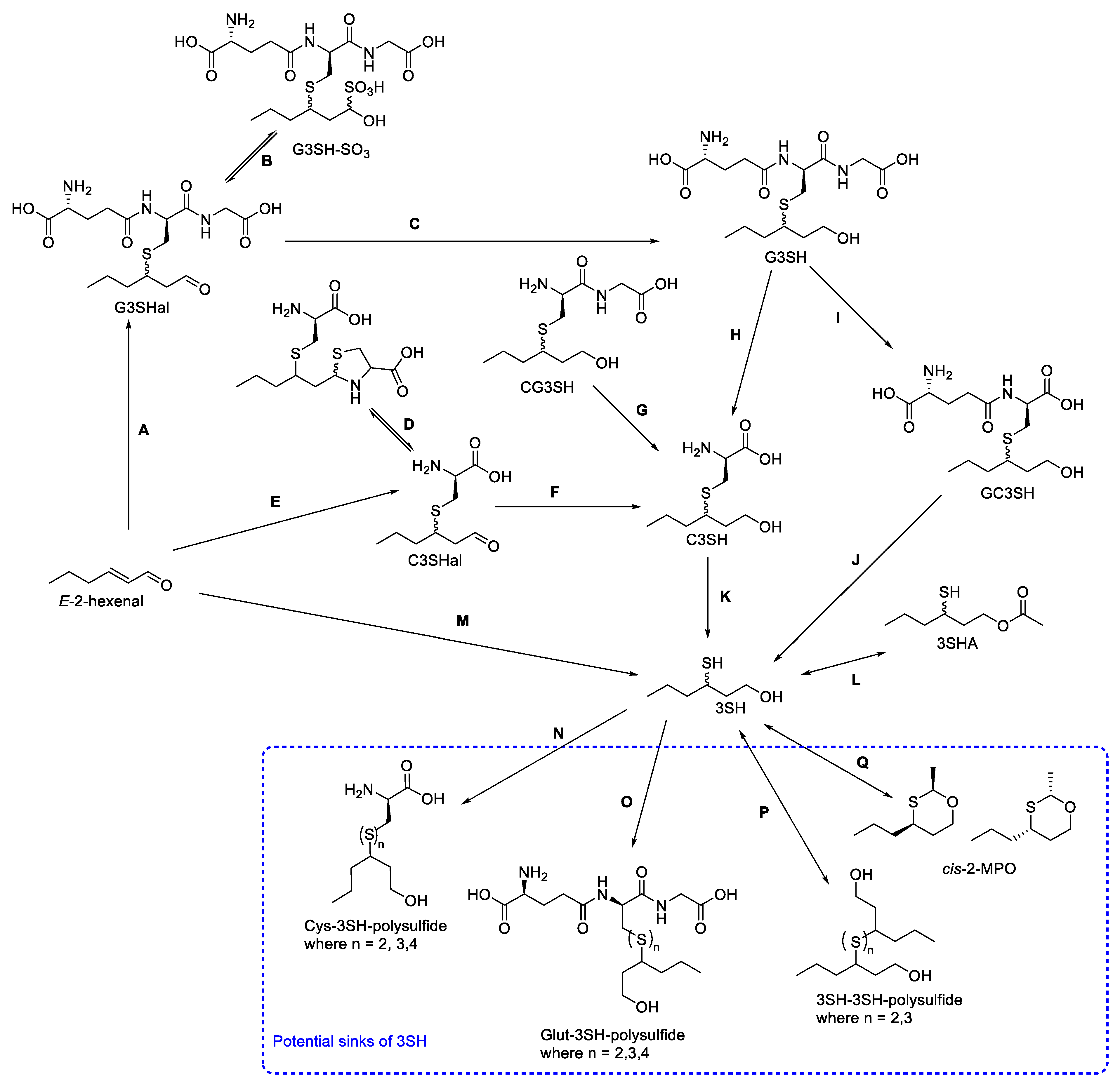
Researchers have used the above analytical methods to widely explore the biogenesis of the volatile thiols 3SH and 3SHA in wines, with a focus on C3SH and G3SH. However, ourthe current understanding of this pathway from these precursors only explains ~50% of the 3SH produced in the finished wine [18,66]. Several studies have shown little correlation between the precursor content of the must and the thiol content of the finished wines [67]. The juice composition strongly influences the release of thiols from their precursors [68], and there are also alternative sources and sinks for 3SH, some confirmed, and others proposed (Scheme 6Scheme 2).Scheme 62. Summary of the current understanding of 3SH formation in enological matrices, as well as key potential sinks of 3SH. Adapted from: A: [69], B: [44,69], C: [70], D: [69], E: [69], F: [69]; G: [66], H: [55], I: [66], J: [66], K: [22,71], L: [72], M: [73], N: [74], O: [74], P: [74], and Q: [75].
Given the impact of many other components of grape juice on precursor consumption and thiol release [68,77], it is unsurprising that studies using grape juice are likely to provide the most insight into 3SH biogenesis. Many of the stable-isotope-labeled isotopomers of thiol precursors can be spiked into samples of real grape juice, either to confirm the nature of these compounds as precursors, or to assess the conversion in real samples. Spiking real grape juice with C3SH-d8 allowed Subileau et al. [56] to gain important insight into the biogenesis of 3SH, in that neither C3SH nor E-2-hexenal were the major sources of 3SH under the fermentation conditions trialed. This is another situation where the location of the incorporation of the labels is important; if the labels can be lost during tautomerism or reaction with components within the wine matrix or are not retained when 3SH is cleaved from the rest of the precursor molecule, the isotopic labels cannot be tracked through to the finished thiols. Bonnafoux et al. [66] found that the potential loss of deuterium labeling during fermentation experiments resulted in an inability to quantify some of the thiols that were produced at extremely low levels, emphasizing the potential for labeling with stable isotopes other than deuterium. Additionally, spiking experiments with isotopologues of thiol precursors can impede the analysis of samples, requiring either an alternative IS or reliance on a different quantification method, such as SA [56,66].
4.1. Aldehyde Conjugate Precursors
Fermentation spiking experiments with stable-isotope-labeled E-2-hexenal led to the initial identification of 3S-glutathionylhexanal (G3Shal) in oenological samples as the stable-isotope-labeled version [70]. The existence of this compound was predicted, as the chemical addition of sulfur-containing compounds to the 1-hexanol moiety can only occur for the α,β-unsaturated carbonyl compound, E-2-hexenal. Although the enzymatic addition of glutathione may occur, it has also been demonstrated that the chemical addition of glutathione to 2-hexenal (Scheme 6 step A) occurs in grape-juice-relevant conditions [69]. G3Shal has been identified in grape juice, and even post-fermentation in finished wines [44,69,82]. One characteristic of aldehydes is their tendency to react with bisulfite or sulfur dioxide to form the corresponding sulfonic acids [83,84,85]. In fact, it has long been suggested that most aldehyde-containing compounds found in wine exist in an equilibrium between the carbonyl and sulfonic acid forms (such as the equilibrium shown in step B of Scheme 6) [84]. As such, it is unsurprising that the first identification and isolation of G3Shal from grape juice also reported the presence of G3SH-SO3, the bisulfite adduct of G3Shal [44].