Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Jiajia Li and Version 3 by Camila Xu.
Integrin β1, also known as CD29, is a human protein-coding gene with a full length of 58048bp. Located on human chromosome 10p11.2 with a total of 18 exons, it has three transcript variants named transcript variants 1A, 1E, and 1D. As the most common β subunit of the integrin family, integrin β1 has been proved to be closely related to the vascular invasion, distant metastasis, and survival of pancreatic cancer (PC) patients, and treatment targeting integrin β1 in PC has gained initial success in animal models.
- integrin β1
- pancreatic cancer
- signaling pathways
1. Introduction
Pancreatic cancer (PC) is the fourth leading cause of cancer-related death worldwide and is characterized by rapid progression, high invasiveness, and resistance to chemotherapeutic agents [1][2][1,2]. More than 80% of the PC lesions have invaded surrounding lymph nodes and might even develop distant metastasis [3]. In theory, the possibility of surgical resection in these patients has been lost and only chemotherapy and palliative care are possible. Due to the complex anatomical position of the pancreas, PC surgery is often more complex than other tumors and cannot completely remove the lesions [4]. Additionally, even if the patients undergo surgical treatment, the postoperative recurrence rate is high, and the median survival time after radical surgery is only 18 months [5]. Despite advances in PC diagnosis and combination therapy of PC over the years, the overall 5-year survival rate remains below 5%, and the median survival time is only close to 6 months [6][7][6,7]. Therefore, targeting the invasion and metastasis of tumor cells is a cutting-edge research area and an urgent issue in PC.
2. An Overview of Integrin β1
Integrin β1, also known as CD29, is a human protein-coding gene with a full length of 58048bp. Located on human chromosome 10p11.2 with a total of 18 exons, it has three transcript variants named transcript variants 1A, 1E, and 1D. Transcript variant 1A has a full length of 3735bp with 16 exons, and encodes a protein of 798 amino acids; transcript variant1E has a full length of 3794bp and encodes a protein of 798 amino acids; transcript variant1D has a full length of 3739bp and encodes a protein of 801 amino acids [8][57].
As the most common β subunit of the integrin family, integrin β1 is currently known to bind to different α subunits to form 11 different integrins. Among these, integrin β1 forms integrins very late antigen (VLA)-1 to 6, α7β1, α8β1, and αvβ1 (vitronectin receptor, VNR) with integrins α1 to α8 and αv subunits, respectively, constituting the VLA family of integrins, which is widely distributed throughout the body [9][58]. A series of studies have unveiled that integrins composed of β1 subunit play a key role in maintaining the stemness property of tumor cells as well as promoting tumor metastasis and chemotherapy/radiation resistance by participating in the transduction of various intracellular signaling pathways [10][11][12][13][14][15][16][59,60,61,62,63,64,65]. In PC, the β1 subunit has been closely related to the vascular invasion, distant metastasis, and survival of the patients, and treatment targeting integrin β1 in PC has gained initial success in animal models [17][66]. Table 1 summarizes the studies regarding integrin β1 in PC.
Table 1.
Overview of cellular functions of reported β1 integrins in pancreatic cancer.
| No. | Integrin | Year | Cell Lines | Expression | Functions | Mechanism | Model Used | Reference PMID | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Proliferation | Cell Cycle | Apoptosis | Angiogenesis | Adhesion | Migration | Invasion | CSC | Therapy Resistance | Tumor Growth | Tumor Metastasis | ECM Remodeling | ||||||||
| 1 | β1 | 2003 | MIA PaCa-2, BxPC-3 | up | + | + | GDNF/integrin β1 | cell | 12883269 | ||||||||||
| 2 | β1 | 2005 | SW1990, Capan-2 | up | + | + | + | GDNF/integrin β1 | cell | 15999351 | |||||||||
| 3 | α6β1 | 2006 | BxPC-3, Capan-2, SW1990 | up | + | + | + | IL-1α/integrin β1 and uPA/uPAR/Ras, ERK | cell | 16504015 | |||||||||
| 4 | β1 | 2006 | Panc-1 | up | + | CCK2/integrin β1/Src, PI3K | cell/animal | 16547500 | |||||||||||
| 5 | β1 | 2006 | Panc-1, BxPC-3 | up | + | integrin β1/FAK/B-catenin phosphorylation/-Lef/Tcf | cell | 16651417 | |||||||||||
| 6 | β1 | 2007 | PATU8902, MIA PaCa-2, Panc-1 | up | + | Cav-1/integrin β1/FAK | cell | 17471232 | |||||||||||
| 7 | β1 | 2007 | Capan-1 | up | + | p16INK4a/glycosylation/integrin β1 maturation | cell | 17535296 | |||||||||||
| 8 | β1 | 2008 | BxPC-3, Panc-1, SW1990 | up | + | integrin β1/ERK1/2 phosphorylation | cell | 17688882 | |||||||||||
| 9 | β1 | 2007 | Panc-1 | up | + | + | Np-1/integrin β1/FAK | cell | 17726369 | ||||||||||
| 10 | β1 | 2008 | Capan-1, Colo-357, AsPC-1, BxPC-3, MIA PaCa-2, Panc-1 | up | + | + | + | - | cell | 18754866 | |||||||||
| 11 | β1 | 2008 | HPDE6 | up | + | CX3CR1/integrin β1/FAK | cell/animal | 18974152 | |||||||||||
| 12 | β1 | 2009 | MIA PaCa-2 | up | + | + | + | cell | 19825166 | ||||||||||
| 13 | β1 | 2010 | bEnd.3, MEF | up | + | + | Fbln5/integrin β1/ROS | cell/animal | 20197418 | ||||||||||
| 14 | β1 | 2011 | FG, Colo-357 | up | + | + | + | + | + | - | cell/animal | 21491421 | |||||||
| 15 | β1 | 2011 | Panc-1, PSN-1, MIA PaCa-2 | up | + | PSC/integrin β1/FAK/radioresistance | cell/animal | 21558392 | |||||||||||
| 16 | β1 | 2011 | Panc-1 | up | + | FAP/ECM/integrin β1 | cell | 21668992 | |||||||||||
| 17 | β1 | 2011 | Panc-1, FG-Met2 | up | + | + | - | cell | 21678462 | ||||||||||
| 18 | β1 | 2011 | T3M4, BxPC-3, COLO-357 | up | + | + | + | + | DNp63a/EGFR, integrin β1/drug resistance | cell | 22053213 | ||||||||
| 19 | β1 | 2012 | Panc-1, AsPC-1 | up | + | integrin β1/Rho | cell | 22232555 | |||||||||||
| 20 | β1 | 2012 | Panc-1 | up | + | - | cell | 22335271 | |||||||||||
| 21 | β1 | 2012 | PT45-P1 | up | + | + | L1CAM/integrin β1/FAK/NF-κB/IL-1β/EMT | cell | 22764136 | ||||||||||
| 22 | β1 | 2013 | Capan-2, FG, Colo-357, Panc-1, Panc1-MUC1 | up | + | + | + | + | + | core 3 synthase/integrin β1/FAK | cell/animal | 23754791 | |||||||
| 23 | α2β1 | 2014 | Panc-1, UlaPaCa | up | + | + | integrin α2β1/FAK | cell | 24201748 | ||||||||||
| 24 | β1 | 2014 | Panc-1, AsPC-1,MIA PaCa-2 | up | + | + | + | + | p53/Myo10/integrin β1/filopodia-inducing | cell/animal | 24487586 | ||||||||
| 25 | β1 | 2014 | AsPC1, BxPC-3, CFPAC-1, Panc-1, SW1990 | up | + | + | + | + | integrin β1/FAK, AKT, and ERK/Gli-1/EMT | cell | 24720337 | ||||||||
| 26 | β1 | 2014 | MIA PaCa-2, BXPC-3 ASPC-1, Panc-1 | up | GD3/integrin β1/FAK/AKT | cell | 24842107 | ||||||||||||
| 27 | β1 | 2014 | PANC-1, MIA PaCa-2 | up | + | + | eEF-2K/TG2/integrin β/Src/uPAR/MMP-2/EMT | cell | 25215932 | ||||||||||
| 28 | α2β1 | 2014 | BxPc-3, Capan-1, Panc-1 | up | + | + | + | integrin β1/FAK/ERK1/2 | cell/animal | 25336636 | |||||||||
| 29 | β1 | 2015 | AsPC-1, Panc-1, MIA PaCa-2 | up | + | + | + | EPAC1/PKC/integrin β1 trafficking and activation | cell/animal | 25385424 | |||||||||
| 30 | β1 | 2015 | AsPC-1, Capan-1, SU.86.86, PANC-1 | up | + | + | + | + | + | - | cell/animal | 25449434 | |||||||
| 31 | β1 | 2016 | ASPC-1, Panc-1, Suit-2 | up | + | + | PHLPP/AKT/integrin β1 | cell | 26760962 | ||||||||||
| 32 | β1 | 2016 | PSC | up | + | integrin β1/ECM/matrix remodeling | cell | 27170254 | |||||||||||
| 33 | β1 | 2016 | MIA PaCa-2, AsPC-1 | up | + | integrin β1/Cdc42, AKT | cell | 27289231 | |||||||||||
| 34 | β1 | 2017 | MIA PaCa-2, AsPC-1, BxPC-3, Panc-1, Capan-2,SW1990 | up | + | + | REGF receptor, neuropilin-1/integrin β1/Src-AKT bypass signaling | cell/animal | 27797376 | ||||||||||
| 35 | β1 | 2017 | Panc-1, L3.6pL, MIA PaCa-2 | up | + | + | NR4A1/p300/Sp/integrin β1 | cell | 28418095 | ||||||||||
| 36 | β1 | 2017 | AsPC-1, BxPC-3, CFPAC1, Panc-1 | up | + | Fyn/P21-activated kinase 1/hnRNP E1/the alterative splicing of integrin β1. | cell/animal | 28560430 | |||||||||||
| 37 | β1 | 2017 | BxPC-3, Capan-1, MIA PaCa-2 | up | Y | + | + | + | integrin β1/FAK | cell/animal | 28692661 | ||||||||
| 38 | α2β1 | 2018 | Panc-1 | up | + | + | + | integrin β1/JNK, ERK kinases, Src | cell/animal | 28916526 | |||||||||
| 39 | β1 | 2017 | AsPC-1, BxPC-3, Panc-1 | up | + | + | + | integrin β1/EGFR/ERK/MAPK/EMT | cell/animal | 29072694 | |||||||||
| 40 | β1 | 2018 | PSC | up | + | + | + | + | GAL3/integrin β1/ILK/NF-kB/IL-8 | cell/animal | 29274868 | ||||||||
| 41 | β1 | 2018 | MIA PaCa-2, Capan-1, AsPC-1 | up | + | + | + | + | + | + | MUC4/integrin β1/FAK/ERK | cell/animal | 29777904 | ||||||
| 42 | β1 | 2018 | MIA PaCa-2 | up | + | + | VASP/integrin β1-FAK-YAP1/TAZ | cell/animal | 29872721 | ||||||||||
| 43 | β1 | 2018 | Panc-1, SW1990, MIA PaCa-2 | up | + | + | + | miR-124/β1/phospho-FAK, phosphor-AKT, phospho-EEK1/2 | cell | 29988949 | |||||||||
| 44 | β1 | 2018 | Panc-1 | up | + | integrin β1/Cdc42 | cell | 30241340 | |||||||||||
| 45 | β1 | 2018 | AsPC-1 | up | + | integrin β1/Cdc42/p110b/PI3K | cell/animal | 30243721 | |||||||||||
| 46 | β1 | 2018 | Panc-1, PK59 | up | + | + | H19/integrin β1,CD24 | cell | 30410672 | ||||||||||
| 47 | β1 | 2019 | Capan-1,BxPC-3 | up | + | + | + | integrin β1/FAK/EMT | cell | 30747824 | |||||||||
| 48 | α11β1 | 2019 | myCAF | up | + | + | - | cell | 31159419 | ||||||||||
| 49 | α5β1 | 2019 | MIA PaCa-2, SW1990, CFPAC-1, PANC-1, AsPC-1, BxPC-3, Panc 03.27 | up | + | + | TGF-β/TFEB/RAB5A/α5β1 endocytosis | cell/animal | 31387632 | ||||||||||
| 50 | β1 | 2019 | MIA PaCa-2 | up | + | integrin β1/c-Myc degradation | cell | 31452837 | |||||||||||
| 51 | β1 | 2019 | SW1990, AsPC-1, Panc-1, BxPC-3 | up | + | + | + | miR-760/MOV10/integrin β1 | cell | 31693728 | |||||||||
| 52 | α3β1 | 2020 | AsPC-1, MIA PaCa-2 | up | + | + | + | + | ZIP4/ZEB/α3β1/JNK/ENT1/drug resistance | cell/animal | 31711924 | ||||||||
| 53 | β1 | 2020 | Panc-1, BxPC-3, MIA PaCa-2 | up | + | HLA-B/integrin β1 | cell | 32194036 | |||||||||||
| 54 | β1 | 2020 | PANC-1 | up | + | + | integrin β1 and Heparan Sulfate Dual-Targeting/YAP | cell | 32266811 | ||||||||||
| 55 | β1 | 2020 | iKras*p53* PC cells | up | + | integrin β1/Kras | cell | 32636409 | |||||||||||
| 56 | β1 | 2020 | Panc-1 | up | + | + | integrin β1/FAK, AKT, ERK1/2, NF-κB | cell | 33086527 | ||||||||||
| 57 | β1 | 2022 | Panc-1 | up | + | + | + | FxOH/integrin β1/FAK, paxillin, FYN, AKT, PPARγ | cell | 33590779 | |||||||||
| 58 | β1 | 2021 | Panc-1 | up | + | mi-16/integrin β1/PI3K/AKT | cell | 33591944 | |||||||||||
| 59 | β1 | 2021 | Panc-1, MIA PaCa-2 | up | + | + | + | hERG1/integrin β1 complex/AKT, HIF-1α | cell/animal | 34045227 | |||||||||
| 60 | β1 | 2022 | MIA PaCa-2 | up | + | - | cell | 34481933 | |||||||||||
| 61 | β1 | 2021 | MIA PaCa-2 | up | + | integrin β1/kindlin-2/TGF-β receptor 2/Smad2/3 | cell | 34638957 | |||||||||||
| 62 | β1 | 2022 | adipose-derived mesenchymal stem cells | up | + | + | + | Mucin 5AC/CD44-integrin β1/Rac1 | cell/animal | 35219699 | |||||||||
| 63 | β1 | 2021 | CF Pac-1, SW1990 | up | + | RAB5A/integrin β1/Cdc42 | cell | 33341673 | |||||||||||
“+” means that the integrin plays a role in related cancer cell biological functions. * iKras*p53* PC means PC cells derived from transgenic mice “iKras*p53* mice”.
3. Roles of Integrin β1 in the Malignant Behaviors of PC
3.1. Integrin β1 and Proliferation-Related Signaling
Integrins are critical for cell proliferation under physiological and pathological conditions [18][67]. As for the behind mechanism, interactions between integrins and growth factor receptors (GFR) have drawn increasing attention these years. In normal cells, diverse receptors including EGFR, cMet, PDGFR, and VEGFR can interact with integrins to activate cell proliferation activity. Previous studies found that integrins could directly activate EGFR in normal endothelial cells independent of EGF. Experiments in liver regeneration demonstrated that the downregulation of the β1 subunit in hepatocytes decreased c-Met and EGFR phosphorylation, thereby inhibiting cell proliferation [19][20][68,69]. In tumor cells, integrins also play a part in cell proliferation by interacting with GFR [21][70]. In vivo/in vitro experimental studies discovered the interaction between EGFR and integrin α5 as well as integrin β1 in tumors [22][23][71,72]. Studies in hepatoma have found that the cellular macromolecular protein cysteine-rich protein 61 (CYR61/CCN1) can stimulate the accumulation of reactive oxygen species (ROS) in cells by interacting with integrin α6β1, thereby inhibiting the activation of the EGFR signaling pathway and the proliferation of hepatoma cells; in addition, using siRNA to interfere with the expression of the integrin β1 subunit could effectively suppress the viability of hepatoma cells [24][25][73,74]. Regarding PC, overexpression of integrin β1 and the downstream Src-AKT activation have been reported. This triggers an EGFR ligand-independent proliferation signaling, bypassing the EGFR-blocking effect, and mediates resistance to the anti-EGFR monoclonal antibody, cetuximab. Knockdown of integrin β1 or inhibition of Src or AKT can successfully re-sensitize cetuximab-resistant (CtxR) PC cells to cetuximab. The researchers then discovered that neuropilin-1 (NRP1) physically interacted with active integrin β1, but not the inactive one on the cell surface. They generated an EGFR and NRP1 dual targeting antibody, Ctx-TPP11, to simultaneously inhibit active integrin β1-driven signaling and suppress EGFR signaling. Further experiments proved the efficacy of Ctx-TPP11 on the inhibition of PC proliferation, both in vitro and in vivo. This researchtudy offered an effective therapeutic strategy based on EGFR and integrin β1 dual targeting, which might become a hot topic in PC therapy [26][75].
3.2. Integrin β1 and Tumor Suppressor p53
Numerous studies have reported the inactivation of the tumor suppressor protein p53 in different forms of human cancer [27][76]. It is believed that the leading cause for the inactivation of the wild-type p53 signaling pathway in tumor cells is the deletion mutation of the p53 gene or abnormal upregulation of its inhibitory proteins, among which integrins play an indispensable role [28][29][77,78].
Previous literature demonstrates that tumor cells can suppress p53 activation through integrin α5β1 in response to chemotherapeutic drugs, thereby downregulating the drug sensitivity [30][79]. In breast cancer, inhibiting integrin α2β1 can upregulate the expression of wild-type p53. At the same time, glioma cells can inhibit the expression of wild-type p53 by upregulating integrin α5 to enhance tumor chemotherapy resistance [31][80]. Recent studies have found that the integrin α5β1/AKT/PEA15/caspase8 signaling pathway in glioma can directly regulate the activity of p53. Integrins can also interact with p53 through the downstream protein kinase focal adhesion kinase (FAK), thus inhibiting its activity [32][81]. In PC, integrin β1 is also found to mediate mutant p53-driven cancer invasion, which needs the facilitation of a filopodia-inducing motor protein, Myosin-X (Myo10). Experiments using a Myo10 mutant without the integrin-binding domain revealed that the ability of Myo10 to transport integrin β1 to the filopodia tip is required for invasion. The introduction of mutant p53 promoted Myo10 expression in a mouse PC model, whereas suppression of endogenous mutant p53 attenuated Myo10 levels and cell invasion. These results suggest that cell components that contribute to plasma-membrane protrusions, such as integrin β1, may serve as specialized metastatic engines for mutant p53-driven PC [33][82]. It is believed that activation of p53 function is a vital strategy to intervene in tumor progression [34][83]. The studies mentioned above imply that inhibiting the activity of integrins and their downstream signaling pathways will provide a new approach to activating p53 and will become a novel research orientation in the field of tumor therapy.
3.3. Integrin β1 and Cell Apoptosis
Regulation of the normal cell cycle via interaction of integrins with ECM is crucial in promoting embryonic development and maintaining tissue homeostasis. The imbalance of this interaction and the resulting inactivation of the downstream PI3K/AKT, MEK/ERK, FAK, NF-κB, and ILK signaling pathways could trigger abnormal cell apoptosis [35][84]. During tumor metastasis, due to disengagement from the interaction with ECM, cancer cells exhibit an enhanced ability to resist apoptosis by reprogramming the expression of integrins [36][85]. In hepatoma cells, upregulation of miR-26a can inhibit the expression of integrin α5 and promote tumor apoptosis [37][86]. It has been reported that melanoma cells express the matrix metalloproteinase inhibitor 1 (TIMP1) to resist apoptosis by forming complexes with CD63 and integrin β1 [38][87]. In breast cancer, upregulation of integrin α6β1 can decrease the expression of non-receptor tyrosine kinase FER in the cytoplasm, thereby impairing the ability to resist apoptosis [39][88]. In addition, vacuolar–ATPase inhibitors have been shown to regulate the anti-apoptotic ability of various tumor cells by reducing the activity of integrin β1 [40][51], and the zinc finger transcription factor ZNF304 can enhance the resistance to cell apoptosis by regulating integrin β1 transcription [41][42][89,90]. In PC, integrin β1 is also reported to be involved in apoptosis. Notably, numerous materials have been identified to regulate this process. For example, methylseleninic acid can induce entosis by cell detachment through downregulation of Cdc42 and integrin β1, and fucoxanthinol (FxOH) suppresses apoptosis of PANC-1 cells by upregulating the expression of integrin β1, FAK, paxillin, FYN, AKT, and PPARγ [43][44][91,92].
3.4. Integrin β1 and Angiogenesis
The involvement of integrins in regulating angiogenesis under various conditions has been extensively investigated. In tumor therapy, αvβ3/β5 is the first group of integrins identified to have the function of promoting the growth of new tumor vessels, and its functional antagonist cilengitide is also the first anti-tumor angiogenesis drug used in clinical research [45][93]. Unfortunately, cilengitide failed to improve overall survival in a multicenter, randomized, controlled phase 3 study in glioma [46][94]. Subsequent studies suggest that the antitumor effect of cilengitide is closely related to the time and dose of administration, and different conditions may lead to opposite results [47][95]. α5β1 is another integrin that promotes tumor angiogenesis [21][48][70,96]. Studies on the molecular mechanism behind the β1 subunit regulating angiogenesis found that angiopoietin-2, Arf6, VE-cadherin, and MAP4K4 were involved in activating the β1 signaling pathway, thereby regulating the subsequent angiogenesis process [49][50][51][52][97,98,99,100]. Integrins can accelerate the growth of new blood vessels and enhance the resistance to anti-angiogenic drugs. Bevacizumab, a commonly used anti-tumor angiogenesis drug, has shown promising effects in treating various tumors [53][101]. In clinical practice, however, drug resistance emerged during the treatment of glioma with Bevacizumab. Studies have detected that the expression of integrin α5β1 and its ligand fibronectin in drug-resistant tumor cells is significantly increased, while interfering with the expression of β1 can restore the sensitivity of tumor cells to Bevacizumab and improve the therapeutic efficacy [54][55][102,103]. While anti-angiogenic drugs have not been applied in the treatment of PC, research on the engagement of integrin β1 in PC angiogenesis has also achieved progress. Studies reveal that the loss of integrin β1 binding to fibulin-5 (Fbln5) can inhibit tumor angiogenesis in endothelial cells by increasing the level of ROS, suggesting that blocking Fbln5 function or interaction between Fbln5 and integrin β1 could be an effective anti-tumor strategy, alone or in combination with other therapies [56][104].
3.5. Integrin β1 and Metastasis
Early metastasis is a hallmark of PC pathology. The entire metastatic process involves the following distinct steps: EMT, invasion, intravasation (from primary tumor sites to enter blood vessels or lymphatic vessels), extravasation (from circulation to distant metastasis sites), and colonization to form secondary malignant tumor [57][58][105,106]. During this process, the roles of integrin β1 go all from the beginning to the end [59][60][107,108]. By activating integrin β1, the HGF/c-Met signaling pathway can promote the EMT transformation of gastric cancer [61][62][63][64][109,110,111,112]. Sheng et al. found that EGF simultaneously induced EMT and activated the integrin β1/EGFR-ERK/MAPK signaling pathway in three PC cell lines. This pathway could be further regulated by Calreticulin (CRT). Immunofluorescence showed that CRT was co-stained with pEGFR1173, fibronectin, and integrin β1 in PC cells, and overexpressing CRT reverted the change in EMT-related proteins induced by EGF. These results indicate a crucial function of the integrin β1/EGFR-ERK/MAPK axis signaling pathway in the EMT of PC [65][113].
Early studies show that tumor cells degrade and remodel the ECM by regulating the expression of matrix metalloproteinases (MMPs) through integrin signaling, thereby promoting invasion [66][67][114,115]. In PC, such effects of integrin β1 are executed by MMP-2. Eukaryotic elongation factor-2 kinase (eEF-2K) is an atypical kinase that is highly upregulated in PC cells. Researchers found that downregulation of eEF-2K impaired the invasion of PC cells and significantly decreased the expression of tissue transglutaminase (TG2), a multifunctional enzyme implicated in the regulation of cell attachment, motility, and survival. These alterations were associated with reductions in β1 integrin/uPAR/MMP-2 expressions and suppression in Src activity. Meanwhile, the induction of EMT biomarkers was also compromised by this axis, as demonstrated by the alterations of the zinc finger transcription factors, ZEB1/Snail, and the tight junction protein claudins. Therefore, the β1 integrin/Src/uPAR/MMP-2 signaling pathway represents a novel potential therapeutic target for PC invasion and EMT [68][116].
Apart from facilitating invasion, matrix proteolysis is also engaged in tumor cell intravasation. The behind mechanism involves the production of growth factors and cytokines, which stimulate neo-angiogenesis [69][117]. After circulation in the blood, the next critical step for tumor cells is extravasation. Integrins expressed on both cancer cells and endothelial cells have implications in extravasation. It has been illustrated that endothelial integrin α5 can bind to neuropilin 2 (NRP2), a multi-functional non-kinase receptor for diverse growth factors expressed on cancer cells, mediating extravasation. In the mouse PC model, by interacting with integrin α5 on the endothelial cell, the PC cell can bind to the endothelium and accomplish vascular extravasation [70][118]. Regarding colonization after extravasation, research shows that blockage of activated integrin α5β1 inhibits both lung and bone colonization of breast cancer cells [71][119]. Although similar experiments in PC have not been reported, these studies demonstrate that integrins, especially integrin β1, are in close relationship with tumor metastasis, and, therefore might become a critical target for suppressing PC progression.
3.6. Integrin β1 and Tumor Microenvironment (TME)
As cancer develops, it causes alterations in the surrounding tissue to create a favorable tumor microenvironment (TME) for its successful growth. It mainly includes ECM, surrounding blood vessels, immune cells, fibroblasts, and various signaling molecules [72][120]. It is currently believed that integrins specifically expressed on the cell surface and the corresponding composition of ECM in the tumor microenvironment are the key factors that determine the distant metastasis of tumor cells [73][121]. For example, liver metastasis of colon cancer depends on whether tumor cells express integrins α2β1 and α5β1 that facilitate cell survival in the liver microenvironment [74][122]. Similarly, cancer cells metastasize to the bone via the expression of integrins αvβ3, α2β1, and α4β1 that bind to specific ligands in the bone ECM [75][123]. Pancreatic stellate cells (PSCs) are the most abundant stromal cell types in PC. They are a major source of tumor-associated fibroblasts (CAFs) that can be activated through growth factors secreted by cancer cells. Collagen type V, expressed by PSCs, can affect the malignant phenotype of various PC cell lines, and stable downregulation of collagen type V in PSCs could reduce metastasis in a PC mouse model. This was further shown to be mediated by β1 integrin signaling, since pharmacological and antibody-mediated inhibition of β1 integrin signaling abolished collagen type V-induced effects on PC cells [76][124]. Integrins β1 are also expressed in CAFs. Studies on non-small cell lung cancer demonstrate that knocking out integrin α11β1 in CAFs can interrupt the interaction between tumor cells and CAF and ultimately block the distant metastasis of lung cancer [77][125]. In PC, galectin 3 (GAL3), a β-galactoside-specific lectin, contributes to PC development by stimulating IL8 transcription through integrin β1 on PSCs, further activating NF-κB through integrin-linked kinase (ILK). Thus, inhibiting integrin β1 expression on PSCs can potentially block PC growth [78][126]. Regarding immune response, it has been illustrated that upregulation of integrin αv expression can lower the sensitivity of tumor cells to immune attack caused by chemotherapeutic drugs [79][80][127,128]. Additionally, through the synergistic effect of integrin α5β1 and the extracellular matrix tenascin C, tumor cells can avoid the infiltration and attack of the surrounding immune cells [80][128]. In vivo experiments found that upregulation of the expression of integrin α4β1 can stimulate the activation and infiltration of T lymphocytes in the tumor tissues, thereby restraining tumor growth [81][129]. Recent research has found that normal immune cells can promote tumor metastasis in a specific environment, and it is speculated that the mechanism could be that tumor cells might enhance the invasion and metastasis activities by interacting with integrin αMβ2 contained in exosomes secreted by immune cells [82][130]. Research on the regulation of PC tumor immunity by integrin β1 is still lacking, but this may hopefully become a future direction for further investigation.
3.7. Integrin β1 and CSCs
Recently, a subpopulation of cells with self-renewal and differentiation abilities, termed CSCs, has been described and is assumed to be the driver for malignant characteristics by engaging in the processes of tumor growth, metastasis, and drug resistance [83][84][85][131,132,133]. CSCs are often identified with an expression of stemness markers including CD24, CD44, Nanog, CD133, Sox2, Sox9, essential specific antigen (ESA), and Kruppel-like factor 4 (KLF4) [86][87][88][134,135,136]. Integrins have been illustrated to play a pivotal part in cancer initiation, progression, and differentiation, indicating their contribution to CSC properties in diverse human cancers, including PC [89][137]. Barnawi and his colleagues analyzed the expression profiles of β1 integrin in 530 breast cancer patients and reported a correlation between β1 integrin and fascin expression; further research demonstrated that fascin facilitated the abilities of adhesion, self-renewal, and chemoresistance in breast cancer cells through β1 integrin [90][91][138,139]. In mice lacking β1-integrin function, complete inhibition of tumorigenesis was observed; in the mammary gland, tissue-specific loss of function of β1 integrin can effectively abrogate the generation and proliferation of CD24hiCD29loCD61hi cancer cells [92][93][140,141]. Studies in squamous cell carcinoma reveal that α6hiβ1hi cells can initiate secondary tumors while those with α6loβ1lo expression cannot, providing evidence for integrin β1-mediated CSC properties [94][142]. In PC, researchers isolated CD24+CD44+ stem-like cells from the PANC-1 cell line and proved increased invasion ability of these cells compared to CD24-CD44+ cells. Using lectin microarray and nano LC-MS/MS, they identified upregulated integrin β1 expression in CD24+CD44+ stem-like cells [95][143]. Mechanistically, PC cells can activate CAFs and increase collagen synthesis, which further leads to enhanced PC self-renewal and migration, as well as increased frequency of CSCs through FAK activation. Inhibition of the integrin β1/FAK signaling in PC cells significantly blocked the impact of CAFs on clonogenic growth [96][144]. Another research work reports that pancreatic CSCs express elevated aldehyde dehydrogenase (ALDH), which are associated with metastatic property [97][145]. β1 integrin–FAK expression was enriched in these ALDH+ CSCs, and further FAK inhibition abrogates clonogenic PC growth in vitro and in vivo [98][146]. These findings demonstrate that β1 integrin enhances CSC properties and promotes tumor initiation, self-renewal, and metastasis through FAK signaling. Therefore, targeting integrin β1 may potentially be applied as a potent approach for PC treatment to restrain CSC survival and aggressiveness.
3.8. Integrin β1 and Therapy
Surgical resection remains the preferential approach for PC in the early stages. In contrast, for advanced PC, radiotherapy and chemotherapeutic agents, including gemcitabine (Gem), nab-paclitaxel, 5-fluorouacil (5-FU), and FOLFIRINOX, are generally recommended as adjuvant options. Despite the great progress made in these strategies, the overall survival (OS) of PC patients is still unsatisfactory due to the generation of chemoresistance or radioresistance [99][147]. Apart from actions on tumor pathogenesis and progression, increasing numbers of data show that integrins also play important roles in resistance to treatment. Patients with higher levels of integrin β1 tend to be more resistant to chemotherapy and display a worse clinical outcome [100][148]. We will Vdiscuss various factors that are more or less involved in integrin β1-mediated therapeutic resistance will be discussed.
The extensive desmoplastic reaction is a prominent pathological feature of PC and shapes a physical barrier for drug delivery. Under the control of growth factors secreted by PC cells, PSCs can be activated and are responsible for dense ECM deposition, which, in turn, regulates resistance to standard therapies through interaction with tumor cells based on various adhesion molecules, with integrins being the largest family [101][102][103][149,150,151]. A previous study demonstrated that under treatment with 5-FU and Gem, PC cells cultured on collagen V-coated plates exhibit significantly increased survival rates compared to the controls, which can be reversed by inhibiting the integrin β1 signaling pathway [76][124]. In 95% of PC cases, activating mutations in the KRAS oncogene are detected, but clinical agents that directly target mutant KRAS are, so far, not available. Nevertheless, inhibition of downstream effectors, including the MAPK signaling pathway and PI3K signaling pathway, has received increasing attention these days [104][105][152,153]. In a 3D culture model of PC, MEK inhibition induced apoptotic lumen formation, a single-layered cluster with the cells at the periphery of the cluster displaying resistance to MEK inhibition while the cells in the interior layers undergo apoptosis. Following administration of the integrin β1 neutralizing antibody, the cells in the matrigel matrix were scattering, and survival in the context of MEK inhibition significantly decreased [106][154]. Taken together, these data suggest the pivotal role of integrin β1 signaling in the treatment resistance of PC induced by interaction with ECM.
Cumulative evidence supports that integrins can also contribute to drug resistance by interacting membrane proteins other than ECM. For instance, integrin β1 and Caveolin-1(Cav-1), a cell membrane component protein at Caveolae, co-participate in cell motility, invasion, and chemoresistance in lung cancer as well as PC [107][108][155,156]. Notably, the radiosensitivity of PC cells was enhanced, and integrin β1 expression was significantly reduced after Cav-1 silencing [109][110][157,158]. Additionally, integrins can also crosstalk with GFR and transactivate RTK signaling, even in the absence of growth factor ligand, which indicates that integrin signaling has a relationship with acquired resistance to molecularly targeted agents such as Cetuximab, which was discussed earlier in this manuscript [102][150] [111][159]. Regarding the crosstalk mechanism between EGFR and integrins, FAK plays a prominent role in many similar signaling pathways that integrins share with EGFR. At the molecular level, FAK is phosphorylated and activated at distinct domains interacting with various cytoplasmic proteins such as paxillin, Src, PI3K, Grb2, and many others, initiating several downstream signaling pathways [112][113][114][160,161,162]. It is worth noting that through the assembly of FAK-Src-p130Cas complex, the JNK signaling pathway can be activated, leading to negative regulation of equilibrative nucleoside transporter 1 (ENT1) [114][115][162,163]. The ENT1 is well known for mediating gemcitabine intracellular transport and resistance in humans and has, therefore, been proposed as an attractive potential prognostic biomarker for gemcitabine response in PC [116][117][118][164,165,166]. A recent study showed that increased α3 and β1 expression and subsequent activation of integrin α3β1 signaling via JNK inhibit the expression of ENT1, which reduced gemcitabine uptake and accumulation into PC cells [119][167].
Apart from drugs, integrin β1 has also been reported to be involved in cell-adhesion dependent radioresistance via direct contact between PSCs and PC cells. Mantoni et al. demonstrated, for the first time, that PSCs promote radioprotection of PC cells under a direct coculture condition rather than a conditioned medium from PSCs, which is attributed to the integrin β1-FAK signaling activation [120][168]. A further study, conducted by Mohamed et al., also confirmed the role of FAK signaling in improving PSCs-dependent radiosensitivity of cancer cells. The results showed that the FAK–tyrosine kinase inhibitor, VS-4718, can sensitize PC cells for radiation only in the presence of ECM-producing PSCs. The combination of VS-4718 and radiotherapy significantly reduced the growth of tumor aggregates in the 3D multicellular tumor model [121][169]. These effects may be attributed to impaired DNA repair, arrested cell cycle, and enhanced PC stem cell function [96][121][122][144,169,170]. A schematic diagram of the roles of integrin β1 in the malignant behaviors of PC is shown in Figure 12.
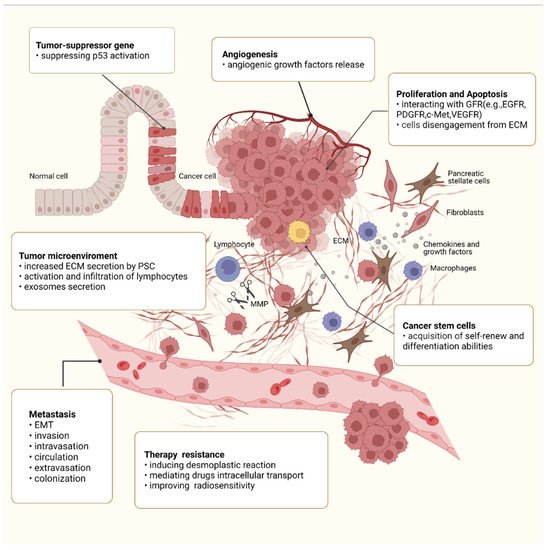
Figure 12. Roles of integrin β1 in the malignant behaviors of PC. Integrin β1 is involved in every step of PC progression and is responsible for regulating malignant cell behaviors such as sustained proliferation, apoptosis resistance, angiogenesis, and migration, as well as CSC property. Integrin β1 also promotes PC metastasis, including EMT, invasion, intravasation, circulation, extravasation, and colonization. In the tumor microenvironment, integrin β1 has been implicated in extensive desmoplastic reaction and regulates the expression of MMPs, the release of GF, and the activation and infiltration of lymphocytes. In addition, integrin β1 also plays an important role in the acquisition of treatment resistance.