Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Conner Chen and Version 4 by Conner Chen.
Cancer is characterized by increased oxidative stress, an imbalance between reactive oxygen species (ROS) and antioxidants. Enhanced ROS accumulation, as a result of metabolic disturbances and signaling aberrations, can promote carcinogenesis and malignant progression by inducing gene mutations and activating pro-oncogenic signaling, providing a possible rationale for targeting oxidative stress in cancer treatment. While numerous antioxidants have demonstrated therapeutic potential, their clinical efficacy in cancer remains unproven.
- reactive oxygen species
- oxidative stress
- cancer therapy
1. ROS Promote Carcinogenesis and Cancer Progression
It was demonstrated that oxidative stress is involved in a wide range of pathologies including cancer, and increased production of ROS are common features of cancer cells. Although high reactive oxygen species (ROS) levels are cytotoxic and may exert anti-tumorigenic effects via oxidative damage and ROS-dependent death signaling, ROS play critical roles during tumorigenesis and cancer development. Here, these contents focus on the pro-tumorigenic role of ROS in malignant progression, which may be addressed with antioxidant therapy. The elevated levels of ROS from altered redox homeostasis contribute to the transformation of healthy cells into cancerous cells and enable their survival through two major mechanisms. The first is that ROS directly oxidize macromolecules, such as nucleic acids, proteins, lipids and glucose, resulting in gene mutation and aberrant inflammation [1]. The second mechanism involves oxidative stress-caused aberrant redox signaling. ROS, particularly hydrogen peroxide (H2O2) and superoxide radical (O2•−), might function as signaling molecules to cause various signaling pathways to go awry and drive cancer progression [2][3] (Figure 1).
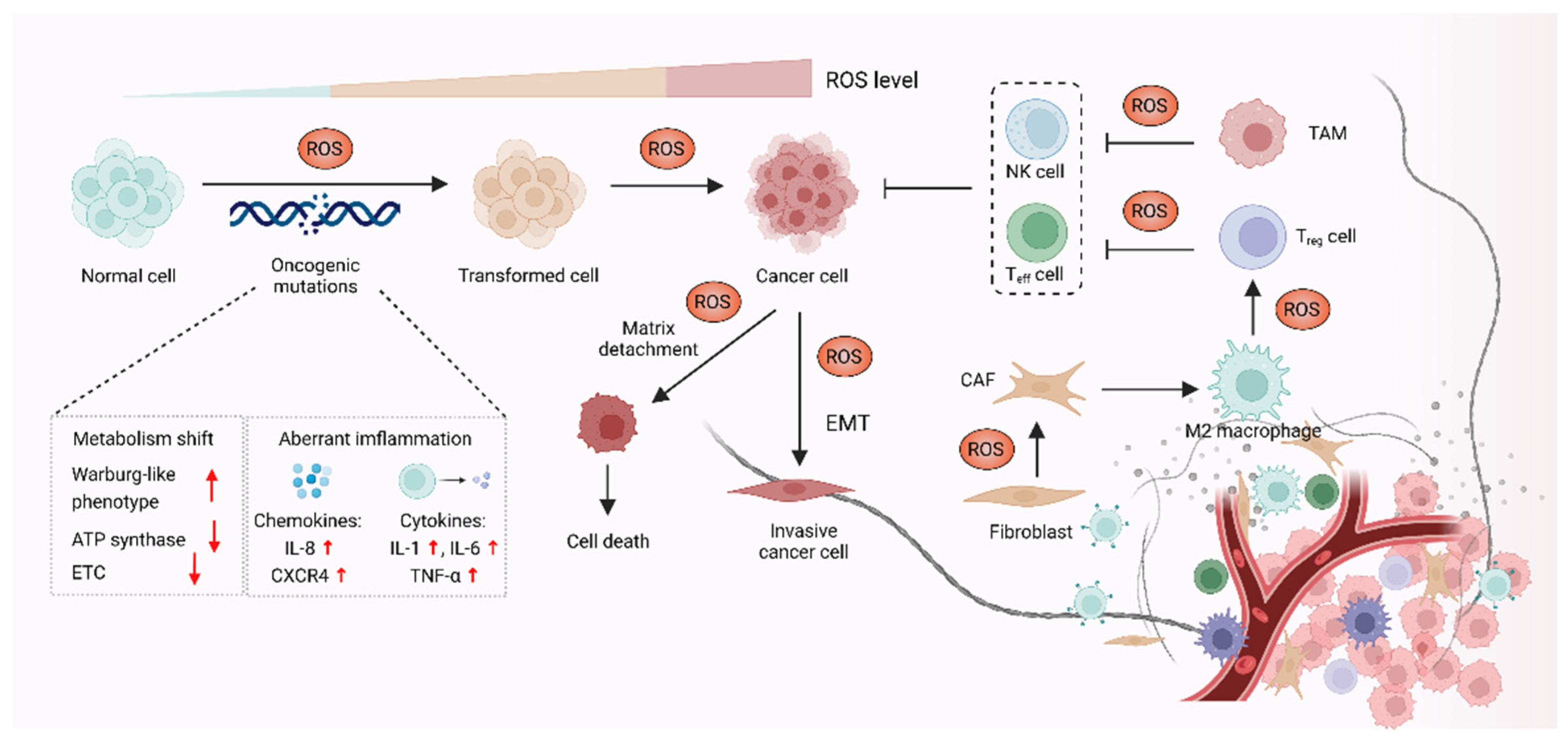
Figure 1. ROS promote carcinogenesis and malignant progression. In the process of carcinogenesis, ROS can contribute to DNA damage, which results in aberrant inflammation and metabolism, leading to oncogenic mutations and cell hyperproliferation. ROS can also act as signaling molecules to enable cancer cells’ survival and cancer progression via epithelial-to-mesenchymal transition (EMT). In addition, ROS might affect stromal cells, such as cancer-associated fibroblasts (CAFs), regulatory T (Treg) cells, effector T (Teff) cells and NK cells in the tumor microenvironment (TME) to promote cancer progression.
1.1. ROS-Mediated Oncogenic Mutations Promote Carcinogenesis
The elevated ROS level functions as a contributor to the malignant transformation of normal cells by inducing mutations in nuclear DNA (nDNA) or mitochondrial DNA (mtDNA), as well as by causing oxidative damage to biomolecules [4][5][6]. Excessive ROS are highly associated with both nDNA and mtDNA mutations, which were reported to result in aberrant inflammation and metabolism, thus promoting malignant transformation [7]. Overproduction of ROS causes nDNA mutation and genetic instability, which further activate multiple oncogenes and lead to abnormal metabolic activity and decreased antioxidant capacity. These events eventually promote the production of ROS in a positive feedback manner [8][9]. Increased ROS was demonstrated to promote chronic inflammation, one of the major causes of cancer, through inducing chemokines such as IL-8 and CXCR4, as well as inflammatory cytokines including IL-1, IL-6 and TNF-α [10][11]. In the context of cancer initiation, mtDNA is also an essential target of ROS, as mtDNA mutation was linked to carcinogenesis [12][13]. Each mitochondrion carries a few dozen mtDNA copies. Increased ROS-induced somatic mutations in mtDNA affect the function of electron transport chain (ETC) and the ATP synthase, which might promote a Warburg-like phenotype shift towards glycolysis. The metabolic shift can shape cell behavior and participate in oncogenic transformation in multiple types of cancer, such as colorectal cancer, lung cancer, gastric cancer, liver cancer and head and neck cancer [14].
1.2. ROS Function as Signaling Molecules to Drive Cancer Progression
In addition to supporting carcinogenesis, ROS were also demonstrated to sustain and accelerate cancer progression via epithelial-to-mesenchymal transition (EMT), which is involved in reprogramming the tumor microenvironment (TME) [15][16]. The TME is affected by ROS through regulating the function of T cells, tumor-associated macrophages (TAMs) and cancer-associated fibroblasts (CAFs) in TME [17]. The TAMs and CAFs promote cell proliferation, angiogenesis, immunosuppression and invasion, thus enabling cancer progression via the reciprocal crosstalk between cancer cells and the TME [18]. Moreover, regulatory T (Treg) cells and cytotoxic CD8+ T cells in TME can suppress effective tumor immunity and contribute to cancer progression, which is associated with poor response to immunotherapy [19][20]. In terms of the role of ROS in TME, H2O2 is thought to function as signaling molecules, which might cause metabolic changes in CAFs, such as altered glucose uptake and mitochondrial activity [21][22]. ROS also contribute to cancer progression by triggering the immunosuppressive properties of TAMs. For instance, mitochondrial ROS activate MAPK/ERK activity, which contributes to the secretion of TNF-α and subsequently promotes cancer invasion [23]. Furthermore, it was also demonstrated that O2•− can suppress T cell-mediated inflammation, thus promoting TAM-mediated immunosuppression and leading to tumor development [24].
2. Antioxidant Therapeutic Strategies in Cancer
Given the important role of ROS in cancer, it follows that modulating ROS levels is a promising anticancer strategy. This may suppress ROS-induced carcinogenesis and cancer progression by inducing oxidative damage and ROS-dependent cell death [7][25]. Therefore, multiple antioxidants and weak pro-oxidants were explored in pre-clinical research and clinical evaluations. Cancer cells can produce excessive ROS through the above-mentioned mechanisms and increased formation of ROS are common features of cancer cells, which makes them more susceptible to a further increase in ROS than normal cells. Therefore, pro-oxidants may function as anticancer agents. For example, it was reported that exogeneous H2O2 can dramatically reduce the survival of MCF-7 cells with PRDX1 knockout, showing the potential of pro-oxidants to promote ROS-mediated cell death [26]. In addition, weak pro-oxidants may also function as important contributors to antioxidant therapy by boosting internal antioxidant capacity. However, treatment with weak pro-oxidants in cancer therapy still needs further investigation. Here, the following contents focus on the antioxidant therapeutic strategies using antioxidants. Overall, antioxidant therapeutic strategies in cancer can be classified as targeting ROS with nonenzymatic antioxidants, including NF-E2 p45-related factor 2 (NRF2) activators [27], vitamins [28][29] (Figure 2) or targeting ROS with enzymatic antioxidants, including NADPH oxidase (NOX) inhibitors [30][31], SOD mimics [32], NAC and GSH esters (Figure 3) (Table 1) [33][34].
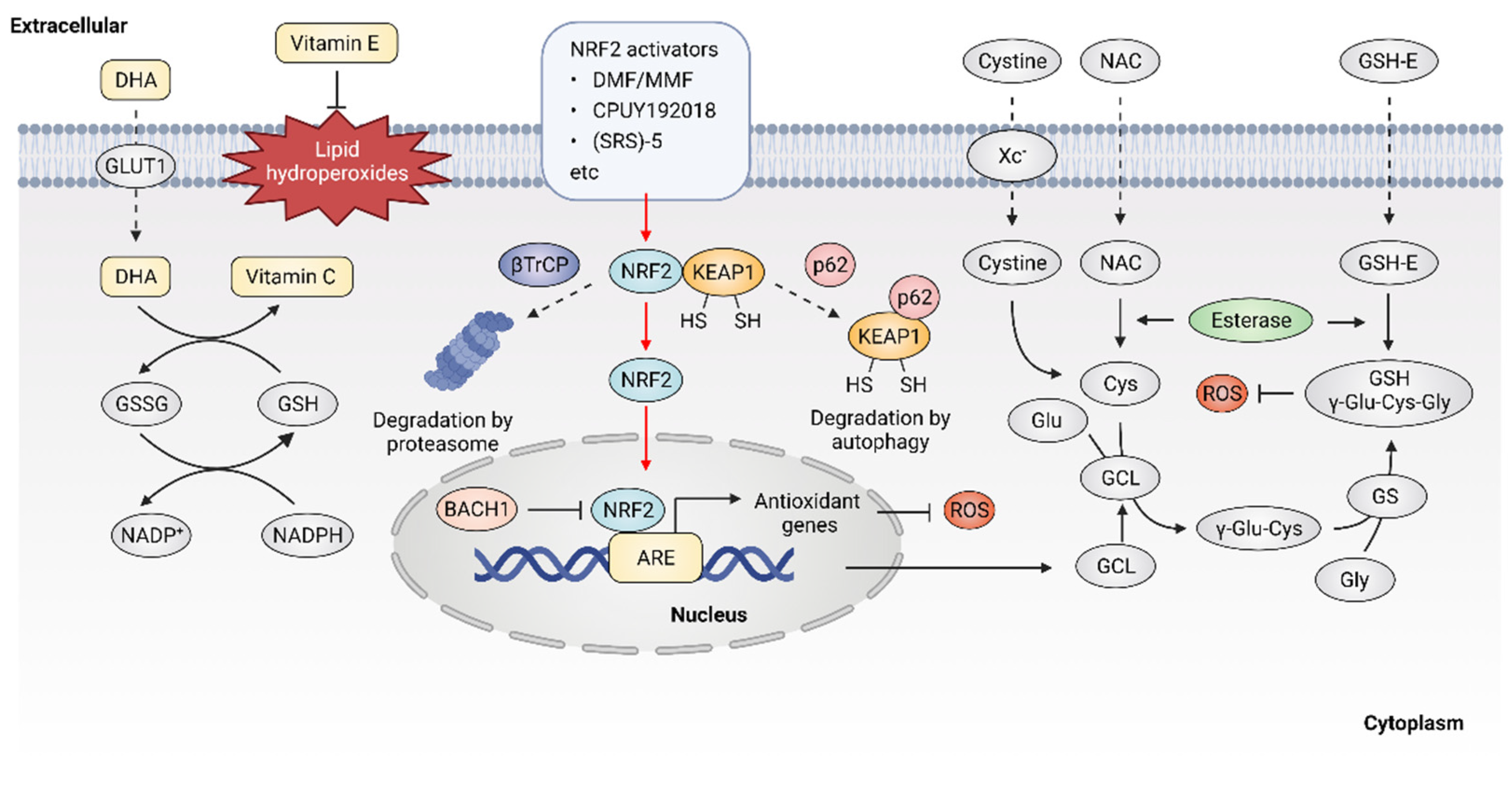
Figure 2. Targeting ROS with nonenzymatic antioxidants. Dehydroascorbic acid (DHA), the oxidized form of vitamin C, is taken up by cells through glucose transporter 1 (GLUT1) and then reduced to vitamin C. Vitamin E is located in cell membranes and defends against lipid hydroperoxides. NRF2 activators may disrupt the KEAP1-NRF2 interaction, leading to the activation of NRF2 downstream antioxidant genes. Glutathione (GSH) is synthesized from cysteine, glutamate and glycine. Exogenous N-Acetyl cysteine (NAC) and GSH esters (GSH-E) supplementation promote GSH production and defense against excessive ROS.
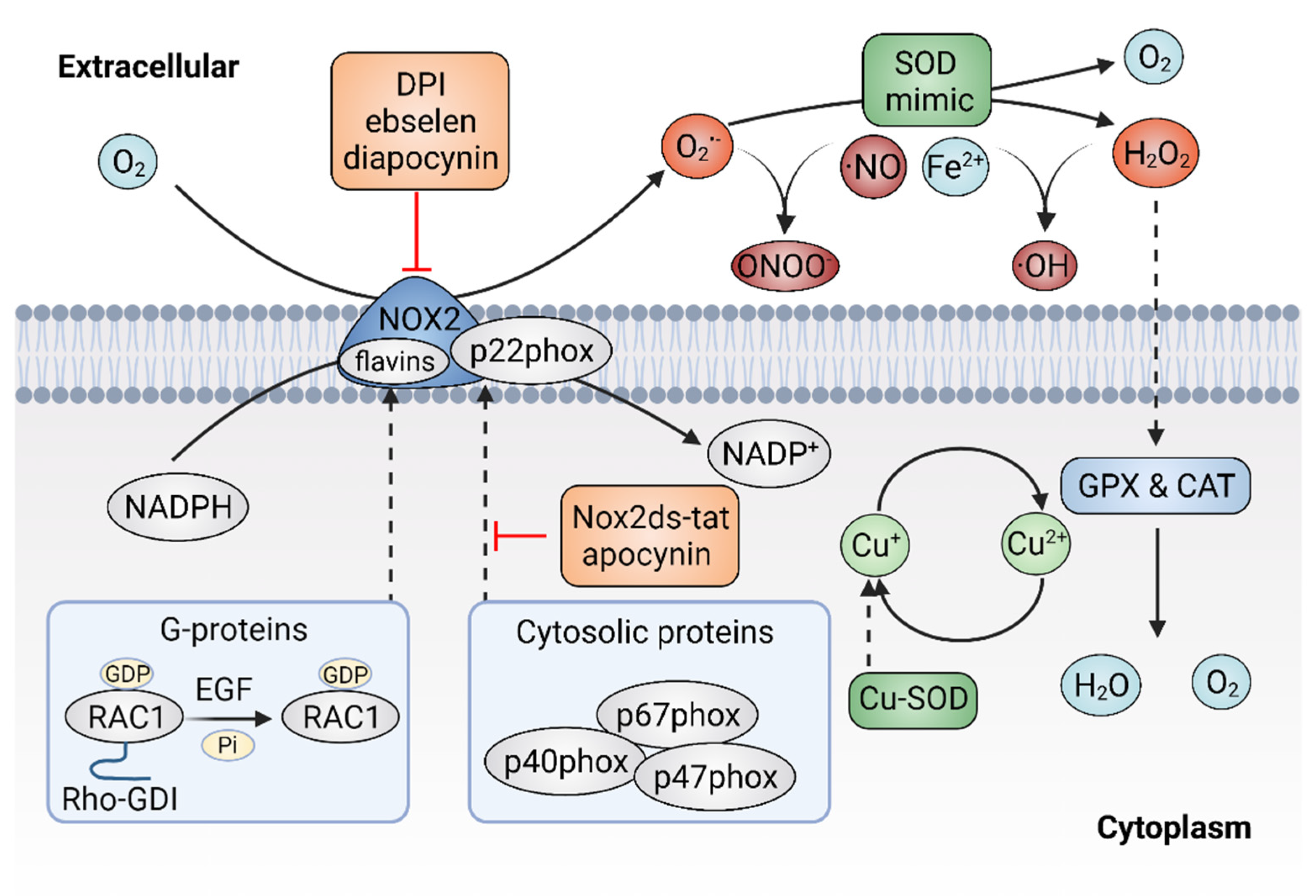
Figure 3. Targeting ROS with enzymatic antioxidants. The inhibitors of plasma membrane NADPH oxidase 2 (NOX2) can prevent the production of superoxide (O2•−) and superoxide dismutase (SOD) mimics might dismutate O2•− to hydrogen peroxide (H2O2).
Table 1. Anticancer antioxidants in clinical trials.
| Antioxidants | Cancer Types | Trial Status | Trial ID |
|---|---|---|---|
| NRF2 activators | |||
| Sulforaphane | Lung cancer | Phase 2 | NCT03232138 |
| Breast cancer | Phase 2 | NCT00982319 | |
| Prostate cancer | Phase 2 | NCT01228084 | |
| Colon cancer | NA | NCT01344330 | |
| HNSCC | Early Phase 1 | NCT03182959 | |
| Resveratrol | Colon cancer | Phase 1 | NCT00256334 |
| Colorectal cancer | Phase 1 | NCT00920803 | |
| Neuroendocrine tumor | NA | NCT01476592 | |
| Breast cancer | NA | NCT03482401 | |
| Multiple myeloma | Phase 2 | NCT00920556 | |
| Quercetin | Prostate cancer | Phase 1 | NCT01912820 |
| Colorectal cancer | NA | NCT00003365 | |
| Pancreatic cancer/NSCLC | Phase 2/3 | NCT02195232 | |
| Curcumin | Breast cancer | Phase 2 | NCT01042938 |
| Colorectal cancer | Phase 2 | NCT02439385 | |
| Prostate cancer | NA | NCT03211104 | |
| Head and neck cancer | Early Phase 1 | NCT01160302 | |
| Pancreatic cancer | Phase 2 | NCT00192842 | |
| Bardoxolone-methyl (CDDO-Me, RTA402) |
Solid tumors/Lymphoid malignancies | Phase 1 | NCT00529438 |
| Pancreatic cancer | Phase 1 | NCT00529113 | |
| Solid tumors/ Lymphoid malignancies | Phase 1 | NCT00508807 | |
| RTA-408 (omaveloxolone) |
NSCLC | Phase 1 | NCT02029729 |
| Breast cancer | Phase 2 | NCT02142959 | |
| Melanoma | Phase 1/2 | NCT02259231 | |
| Dimethyl fumarate | Multiple sclerosis | Phase 3 | NCT02430532 |
| Lymphocytic leukemia | Phase 1 | NCT02784834 | |
| Glioblastoma | Phase 1 | NCT02337426 | |
| Oltipraz | Lung cancer | Phase 1 | NCT00006457 |
| SOD mimics | |||
| GC4419 | Head and neck cancer | Phase 2 | NCT04529850 |
| Pancreatic cancer | Phase 1/2 | NCT03340974 | |
| Squamous cell carcinoma | Phase 1 | NCT01921426 | |
| Head and neck cancer | Phase 2 | NCT02508389 | |
| Metalloporphyrins | Lung cancer | Phase 3 | NCT00054795 |
| NOX inhibitors | |||
| Ebselen (SPI-1005) | Cancer | Phase 1 | NCT01452607 |
| Lung cancer, Head and neck cancer | Phase 2 | NCT01451853 | |
| GSH-related antioxidants | |||
| NAC | Breast cancer | Phase 1 | NCT01878695 |
| Gastric cancer | NA | NCT03238404 | |
| Ovarian cancer | NA | NCT03491033 | |
| Head and neck cancer | Phase 2 | NCT02123511 | |
| Gastrointestinal neoplasms | Phase 2 | NCT00196885 | |
| Bladder cancer | NA | NCT02756637 | |
| Lung cancer | Phase 2 | NCT00691132 | |
| Colorectal cancer | NA | NCT01325909 | |
| NOV-002 | Breast cancer | Phase 2 | NCT00499122 |
| Ovarian cancer | Phase 2 | NCT00345540 | |
| NSCLC | Phase 3 | NCT00347412 | |
| Leukemia | Phase 2 | NCT00960726 | |
| Reduced GSH | Breast cancer | Phase 2 | NCT00266331 |
| Vitamins | |||
| Vitamin C | Ovarian cancer | Phase 2 | NCT00284427 |
| Pancreatic cancer | Phase 1 | NCT00954525 | |
| Prostatic neoplasms | Phase 2 | NCT01080352 | |
| Ovarian cancer | Phase 2 | NCT00284427 | |
| Advanced cancer | Phase 1/2 | NCT01050621 | |
| Solid cancers | Phase 1 | NCT00441207 | |
| NSCLC | Phase 1/2 | NCT02655913 | |
| Head and Neck Cancer | NA | NCT03531190 | |
| Skin cancer | NA | NCT01032031 | |
| Liver cancer | Phase 1/2 | NCT01754987 | |
| Vitamin E | Prostate cancer | Phase 3 | NCT00006392 |
| Colorectal cancer | Phase 1 | NCT00905918 | |
| Head and neck neoplasms | Phase 2 | NCT02397486 | |
| Skin neoplasms | NA | NCT02248584 | |
| Pancreatic neoplasms | Phase 1 | NCT00985777 | |
| Breast cancer | Phase 2 | NCT00022204 |
NA: Not Applicable; HNSCC, head and neck squamous cell carcinoma; NSCLC, Non-small cell lung cancer.
2.1. Targeting ROS with Nonenzymatic Antioxidants
The transcription factor NRF2 was considered as a master regulator of various homeostatic genes that defend against cellular stress, including oxidative stress [35]. Upon exposure to oxidative stress, the transcription factor NRF2 is released from its principal negative regulator Kelch-like ECH-associated protein 1 (KEAP1) and translocates to the nucleus, where NRF2 binds to antioxidant response element (ARE) and promotes the expression of antioxidant genes [36]. High expression of NRF2 was observed in various oxidative stress-related diseases including cancer, especially in NRF2-activated malignant tumors. NRF2 activators were considered as potential agents to prevent carcinogenesis or reverse cancer progression [37]. Five categories of NRF2 activator were developed, the underlying action mechanisms of which include: (1) modification on sensor cysteines of KEAP1, leading to the dissociation between NRF2 and KEAP1 [38][39]; (2) direct disruption of the KEAP1-NRF2 interaction [40]; (3) disruption of the interaction between NRF2 and β-transducin repeat-containing protein (βTrCP), which targets NRF2 for proteasome degradation [41]; (4) sequestration of KEAP1 into autophagosomes by p62 [42]; (5) upregulation of NRF2 protein levels by de novo synthesis that cannot be degraded by KEAP1 [43]; (6) inhibition of the NRF2 transcriptional repressor BTB domain and CNC homolog 1 (BACH1) [44].
The current development of NRF2 activators is mainly based on modifying sensor cysteines of KEAP1 and disrupting the KEAP1-NRF2 interaction. For instance, fumaric acid esters are oral analogs of fumarate that represent a group of NRF2 activators that work by modifying sensor cysteines of KEAP1, among which dimethyl fumarate (DMF) is the most successful example [45]. It was reported that DMF can alkylate Cys151 of KEAP1, leading to the dissociation of NRF2 and KEAP1 [46]. DMF metabolite monomethyl fumarate (MMF) was also demonstrated to react with KEAP1 through Cys151, thereby stabilizing and activating NRF2 [47]. DMF and its major metabolite MMF can reduce inflammatory responses and exhibit a favorable tolerability profile in clinical trials, showing promise for cancer treatment [48]. In addition, compounds that show improved bioavailability compared with MMF, through improving the release rate, were synthesized, such as TFM735, which is reported to activate NRF2 via the Cys151 in KEAP1, leading to the inhibition of IL-6 and IL-17 from peripheral blood mononuclear cells [49]. In addition, nitro fatty acids (NO2-FAs), such as nitro linoleic acid and nitro-oleic acid, are endogenous signaling mediators that react with Cys273 and Cys288 in KEAP1 through nitro alkylation, resulting in the activation of NRF2 and being implicated in anti-inflammatory activities [50]. Recently, the non-covalent NRF2 activators were developed, which directly disrupt the KEAP1–NRF2 protein–protein interaction via a cysteine-independent binding mechanism [51]. For instance, the bis-carboxylic acid compound CPUY192018 is a high-affinity KEAP1 ligand, which promotes the release of NRF2 from KEAP1 and enhances the expression of NRF2-target genes [52]. The sulfonamide-containing compounds were reported to inhibit the KEAP1–NRF2 interaction and enhance the expression of NAD(P)H: quinone oxidoreductase (NQO1), which reduces lung inflammation in animal models [53]. The naphthalene bis-sulfonamide was also reported to promote the expression of NRF2-target NQO1 and protect against dextran sulfate sodium (DSS)-induced colitis [54]. In addition to the above-mentioned compounds, (SRS)-5 and benzene-disulfonamides were also demonstrated to function as potent non-covalent NRF2 activators that disrupt the interaction between KEAP1 and NRF2 [55][56]. Altogether, these compounds are high-affinity ligands for KEAP1 and can directly block the KEAP1–NRF2 interface, thereby activating NRF2 downstream antioxidant genes and protecting cells from oxidative stress. Although current drugs mainly target KEAP1, it is noted that NRF2 might bind to ARE sequences in a KEAP1-independent manner, possibly involving the regulation of transcriptional repressor BACH1 [57]. Therefore, compounds that inhibit the binding of BACH1 to ARE-driven genes, such as HMOX1, were also developed [44]. Presently, more NRF2 activators eliciting beneficial effects are arising. However, treatment with NRF2 activators may inactivate drug-induced oxidative stress that normally would result in cell death. Therefore, it is necessary to monitor their clinical efficacy, given that the activation of NRF2 may contribute to the development of chemoresistance [58][59]. Taken together, NRF2 activators have shown potential for cancer therapy, but further investigations are also needed to demonstrate their clinical efficacy, especially in combination with chemotherapeutic drugs.
NAC is currently one of the most studied antioxidant agents that can be quickly absorbed via the anion exchange membrane and deacetylate to produce cysteine, thus replenishing GSH [60]. NAC can reduce cysteine conjugates and is used therapeutically for many human diseases, including cancers [61]. However, NAC was also reported to increase melanoma cell metastasis in NOD-SCID-Il2rg−/− (NSG) mice [62]. GSH esters, the derivatives of GSH, were developed for GSH supplementation, since GSH cannot be effectively transported into cells and exogenously administered GSH is rapidly cleared in plasma. Ester derivatives of GSH, such as monoethyl (GSH-MEE), diethyl (GSH-DEE), monomethyl (GSH-OMe) and isopropyl esters have shown high efficiency in increasing cellular GSH level [63]. In addition, compared with oral administration, subcutaneous or intraperitoneal injection of GSH esters is more effective in elevating GSH levels in various tissues [64]. However, although the efficacy of GSH esters to alleviate oxidative stress in cells and animal models was demonstrated, clinical trials with GSH ester are still needed.
As the most widely used dietary antioxidants, L-ascorbic acid (vitamin C) and α- tocopherol (vitamin E) are of great interest in cancer therapy [65]. Vitamin C is a type of water-soluble vitamin that cannot be synthesized endogenously in the human body, but can only be provided by dietary supplement, making it an essential nutritional component [66]. Dehydroascorbic acid (DHA), the oxidized form of vitamin C, is absorbed from the renal tubules by renal epithelial cells and functions as a reductant and an enzyme cofactor [67]. It was described that high dose vitamin C shows promising antitumor efficacy in patients with advanced cancer [68][69][70][71]. However, the role of vitamin C in cancer treatment is still controversial, as half of the studies indicate that vitamin C has no significant effect on the incidence and mortality of cancer [72][73][74]. Vitamin E is lipid soluble and mainly localizes to the plasma membrane, where it functions as a ROS scavenger through reacting with free radicals, thus defending against oxidative stress [75]. It was reported that vitamin E only has low toxicity and causes no obvious side effects at high dose intake [76]. However, several animal studies showed that vitamin E supplements might promote carcinogenesis and cancer progression [77]. Overall, the controversial effect of antioxidants on cancer raises significant concerns regarding antioxidant supplements. Therefore, novel strategies are warranted to resolve the double-edged effect of supplemental antioxidants, including vitamin C and vitamin E.
2.2. Targeting ROS with Enzymatic Antioxidants
As mentioned above, the NOX family is a major source of ROS and excessive activation of NOXs can contribute to oxidative stress. Thus, agents that would efficaciously target NOXs to scavenge ROS might hold significant promise for cancer therapy [78]. There are two types of NOXs inhibitors, including peptidic inhibitors and small-molecule inhibitors, both of which are based on the mechanism of inhibiting NOX enzyme activity or suppressing the assembly of the NOX2 enzyme [79]. Small peptide inhibitors of NOX complexes have shown therapeutic potential. The first peptidic inhibitor is Nox2ds-tat ([H]-R-K-K-R-R-Q-R-R-R-C-S-T-R-I-R-R-Q-L-[NH2], also known as gp91ds-tat). Nox2ds-tat was reported to inhibit the assembly of NOX2, a complex that consists of six subunits: the Nox2 subunit (also known as gp91phox); p22phox, and four cytosolic components; p47phox (organizer subunit); p67phox (activator subunit); p40phox, and the small Rho-family GTP binding protein Rac1 or Rac2 [80][81]. Nox2ds-tat selectively blocks NOX2 activity through interrupting the Nox2–p47phox interaction [82]. The inhibitory effects of Nox2ds-tat were demonstrated both in vitro and in vivo. For instance, Nox2ds-tat was reported to inhibit the production of angiotensin II-induced O2•− [83]. Moreover, administration of Nox2ds-tat by subcutaneous infusion significantly attenuated the production of vascular O2•− and subsequent vascular inflammation in angiotensin II-infused rat model [84][85]. In summary, the viability of Nox2ds peptide as a NOX2 inhibitor was demonstrated, which is important for suppressing NOX2 activity and preventing excessive ROS production.
Currently, multiple small-molecule global inhibitors that inhibit NOXs or flavoproteins in general, were synthesized, including diphenyleneiodonium (DPI), ebselen and diapocynin [86]. Among them, DPI is the first identified and commonly used potential inhibitor of NOXs, which inhibits the production of ROS by forming adducts with FAD, potentially contributing to the reduction of ROS and showing anticancer properties in colon cancer cells [87]. However, as a nonselective inhibitor, DPI might target other flavin-dependent enzymes, such as xanthine oxidase and nitric oxide synthase. Ebselen and diapocynin are described as NOX inhibitors but were also previously found to display unrelated effects [88]. Unlike DPI, apocynin specifically prevents the activation of NOX2 by inhibiting the translocation of p47phox, thereby repressing the production of O2− in vitro and exhibiting anti-inflammatory activity in vivo [89]. In addition, other specific NOX inhibitors, were also identified via cellular and membrane assays [90]. For instance, fulvene-5, one of the fulvene derivatives that have a chemical similarity to DPI, could inhibit NOX2 and NOX4 in vitro, as well as block the growth of endothelial cell-derived neoplasia in mice [91]. However, despite the great efforts made by researchers, few NOXS inhibitors have yet reached clinical trials. It remains challenging to identify compounds that target NOX specifically and show a profound impact in alleviating cancer. Much more work is still needed to develop NOX inhibitors for the treatment of oxidative-stress-associated disorders, including cancer.
SOD is a metalloprotein that can efficiently eliminate O2•− with a dismutation mechanism. SOD was developed as a drug known as orgotein, to defend against oxidative stress in mammalian cells [92]. The anti-inflammatory property of orgotein was demonstrated through preclinical and clinical studies [93]. It was also reported that orgotein can effectively prevent or reduce the side effects of radiation therapy in bladder cancer patients [94]. In addition, several types of SOD mimics were synthesized, such as metalloporphyrins, Mn (II) polyamines, Mn (III) salens, Mn (III) corroles and Mn (IV) biliverdins [95][96][97]. Although the rate constants are much lower than the enzymes, SOD mimics appear to be effective in extracellular fluids where the antioxidant enzymes are absent or at deficient concentrations [98]. Moreover, some SOD mimics may act as pro-oxidants rather than antioxidants, thereby activating rather than mimicking SOD [99].
Metalloporphyrins have emerged as the most studied SOD mimics, such as Mn porphyrins. Various Mn porphyrin compounds, including MnTM-2-pYp5+, MnTE-2-pYp5+ and MnTDE-2-ImP5+, have shown high SOD activity that dismutates O2•− to H2O2 [100]. The protective and therapeutic potential of Mn porphyrins were demonstrated in animal models of diseases, including cancers. To date, more porphyrins or porphyrin-based SOD mimics were synthesized with the establishment of the structure–activity relationships between SOD and metal-site redox ability [101]. The Mn (II)-containing penta-aza macrocyclic manganese compound GC4419 (known as avasopasem manganese, AVA) was reported to enhance tumor-killing activity when synergized with radiation in head and neck cancer [102]. In addition, GC4419 can enhance the toxicity of high-dose vitamin C in a H₂O₂-dependent manner, promoting radiation-induced cancer cell killing [103]. Furthermore, GC4419 also exhibits therapeutic potential in the inflammation animal model [104]. Unlike GC4419, the Mn (III)- containing salen complexes, such as EUK-8, EUK-134 and EUK-189, are not specific and have dismutation activity on both O2•− and H2O2, showing protective effects for various types of cancer [105].
In summary, multiple antioxidant therapeutic strategies were developed for cancer treatment, which can be classified into two different categories of groups according to their targets: enzymatic antioxidants and nonenzymatic antioxidants, both of which have shown potential to act as antioxidant drugs in pre-clinical and clinical research.
References
- Zhong, H.; Yin, H. Role of lipid peroxidation derived 4-hydroxynonenal (4-HNE) in cancer: Focusing on mitochondria. Redox Biol. 2015, 4, 193–199.
- Ye, Z.W.; Zhang, J.; Townsend, D.M.; Tew, K.D. Oxidative stress, redox regulation and diseases of cellular differentiation. Biochim. Biophys. Acta 2015, 1850, 1607–1621.
- Parascandolo, A.; Laukkanen, M.O. Carcinogenesis and Reactive Oxygen Species Signaling: Interaction of the NADPH Oxidase NOX1-5 and Superoxide Dismutase 1–3 Signal Transduction Pathways. Antioxid. Redox Signal. 2019, 30, 443–486.
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084.
- Lood, C.; Blanco, L.P.; Purmalek, M.M.; Carmona-Rivera, C.; De Ravin, S.S.; Smith, C.K.; Malech, H.L.; Ledbetter, J.A.; Elkon, K.B.; Kaplan, M.J. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat. Med. 2016, 22, 146–153.
- Ishikawa, K.; Takenaga, K.; Akimoto, M.; Koshikawa, N.; Yamaguchi, A.; Imanishi, H.; Nakada, K.; Honma, Y.; Hayashi, J. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 2008, 320, 661–664.
- Poprac, P.; Jomova, K.; Simunkova, M.; Kollar, V.; Rhodes, C.J.; Valko, M. Targeting Free Radicals in Oxidative Stress-Related Human Diseases. Trends Pharmacol. Sci. 2017, 38, 592–607.
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64.
- Niedernhofer, L.J.; Gurkar, A.U.; Wang, Y.; Vijg, J.; Hoeijmakers, J.H.J.; Robbins, P.D. Nuclear Genomic Instability and Aging. Annu. Rev. Biochem. 2018, 87, 295–322.
- Blaser, H.; Dostert, C.; Mak, T.W.; Brenner, D. TNF and ROS Crosstalk in Inflammation. Trends Cell Biol. 2016, 26, 249–261.
- Gupta, S.C.; Hevia, D.; Patchva, S.; Park, B.; Koh, W.; Aggarwal, B.B. Upsides and downsides of reactive oxygen species for cancer: The roles of reactive oxygen species in tumorigenesis, prevention, and therapy. Antioxid. Redox Signal. 2012, 16, 1295–1322.
- Zhao, M.; Wang, Y.; Li, L.; Liu, S.; Wang, C.; Yuan, Y.; Yang, G.; Chen, Y.; Cheng, J.; Lu, Y.; et al. Mitochondrial ROS promote mitochondrial dysfunction and inflammation in ischemic acute kidney injury by disrupting TFAM-mediated mtDNA maintenance. Theranostics 2021, 11, 1845–1863.
- Kopinski, P.K.; Singh, L.N.; Zhang, S.; Lott, M.T.; Wallace, D.C. Mitochondrial DNA variation and cancer. Nat. Rev. Cancer 2021, 21, 431–445.
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721.
- Koelwyn, G.J.; Quail, D.F.; Zhang, X.; White, R.M.; Jones, L.W. Exercise-dependent regulation of the tumour microenvironment. Nat. Rev. Cancer 2017, 17, 620–632.
- Kubli, S.P.; Bassi, C.; Roux, C.; Wakeham, A.; Gobl, C.; Zhou, W.; Jafari, S.M.; Snow, B.; Jones, L.; Palomero, L.; et al. AhR controls redox homeostasis and shapes the tumor microenvironment in BRCA1-associated breast cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 3604–3613.
- Weinberg, F.; Ramnath, N.; Nagrath, D. Reactive Oxygen Species in the Tumor Microenvironment: An Overview. Cancers 2019, 11, 1191.
- Casey, S.C.; Amedei, A.; Aquilano, K.; Azmi, A.S.; Benencia, F.; Bhakta, D.; Bilsland, A.E.; Boosani, C.S.; Chen, S.; Ciriolo, M.R.; et al. Cancer prevention and therapy through the modulation of the tumor microenvironment. Semin. Cancer Biol. 2015, 35, S199–S223.
- Krejcik, J.; Casneuf, T.; Nijhof, I.S.; Verbist, B.; Bald, J.; Plesner, T.; Syed, K.; Liu, K.; van de Donk, N.W.; Weiss, B.M.; et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood 2016, 128, 384–394.
- La Fleur, L.; Botling, J.; He, F.; Pelicano, C.; Zhou, C.; He, C.; Palano, G.; Mezheyeuski, A.; Micke, P.; Ravetch, J.V.; et al. Targeting MARCO and IL37R on Immunosuppressive Macrophages in Lung Cancer Blocks Regulatory T Cells and Supports Cytotoxic Lymphocyte Function. Cancer Res. 2021, 81, 956–967.
- Druzhkova, I.N.; Shirmanova, M.V.; Lukina, M.M.; Dudenkova, V.V.; Mishina, N.M.; Zagaynova, E.V. The metabolic interaction of cancer cells and fibroblasts—coupling between NAD(P)H and FAD, intracellular pH and hydrogen peroxide. Cell Cycle 2016, 15, 1257–1266.
- Zhu, J.; Thompson, C.B. Metabolic regulation of cell growth and proliferation. Nat. Rev. Mol. Cell Biol. 2019, 20, 436–450.
- Kastl, L.; Sauer, S.; Beissbarth, T.; Becker, M.; Krammer, P.; Gulow, K. TNF-a stimulation enhances ROS-dependent cell migration via NF-?B activation in liver cells. Free Radic. Biol. Med. 2014, 75 (Suppl. S1), S32.
- Li, G.; Liu, D.; Kimchi, E.T.; Kaifi, J.T.; Qi, X.; Manjunath, Y.; Liu, X.; Deering, T.; Avella, D.M.; Fox, T.; et al. Nanoliposome C6-Ceramide Increases the Anti-tumor Immune Response and Slows Growth of Liver Tumors in Mice. Gastroenterology 2018, 154, 1024–1036.e1029.
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709.
- Bajor, M.; Zych, A.O.; Graczyk-Jarzynka, A.; Muchowicz, A.; Firczuk, M.; Trzeciak, L.; Gaj, P.; Domagala, A.; Siernicka, M.; Zagozdzon, A.; et al. Targeting peroxiredoxin 1 impairs growth of breast cancer cells and potently sensitises these cells to prooxidant agents. Br. J. Cancer 2018, 119, 873–884.
- Schmidlin, C.J.; Shakya, A.; Dodson, M.; Chapman, E.; Zhang, D.D. The intricacies of NRF2 regulation in cancer. Semin. Cancer Biol. 2021, 76, 110–119.
- Young, M.R.I.; Xiong, Y. Influence of vitamin D on cancer risk and treatment: Why the variability? Trends Cancer Res. 2018, 13, 43–53.
- Bakalova, R.; Zhelev, Z.; Miller, T.; Aoki, I.; Higashi, T. New potential biomarker for stratification of patients for pharmacological vitamin C in adjuvant settings of cancer therapy. Redox Biol. 2020, 28, 101357.
- Augsburger, F.; Filippova, A.; Rasti, D.; Seredenina, T.; Lam, M.; Maghzal, G.; Mahiout, Z.; Jansen-Durr, P.; Knaus, U.G.; Doroshow, J.; et al. Pharmacological characterization of the seven human NOX isoforms and their inhibitors. Redox Biol. 2019, 26, 101272.
- Liang, S.; Ma, H.Y.; Zhong, Z.; Dhar, D.; Liu, X.; Xu, J.; Koyama, Y.; Nishio, T.; Karin, D.; Karin, G.; et al. NADPH Oxidase 1 in Liver Macrophages Promotes Inflammation and Tumor Development in Mice. Gastroenterology 2019, 156, 1156–1172.e1156.
- Batinic-Haberle, I.; Tovmasyan, A.; Spasojevic, I. Mn Porphyrin-Based Redox-Active Drugs: Differential Effects as Cancer Therapeutics and Protectors of Normal Tissue Against Oxidative Injury. Antioxid. Redox Signal. 2018, 29, 1691–1724.
- Wu, Z.Y.; Kim, H.J.; Lee, J.W.; Chung, I.Y.; Kim, J.S.; Lee, S.B.; Son, B.H.; Eom, J.S.; Kim, S.B.; Gong, G.Y.; et al. Breast Cancer Recurrence in the Nipple-Areola Complex After Nipple-Sparing Mastectomy With Immediate Breast Reconstruction for Invasive Breast Cancer. JAMA Surg. 2019, 154, 1030–1037.
- Wu, J.H.; Batist, G. Glutathione and glutathione analogues; therapeutic potentials. Biochim. Biophys. Acta 2013, 1830, 3350–3353.
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203.
- Dodson, M.; Castro-Portuguez, R.; Zhang, D.D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019, 23, 101107.
- Mills, E.L.; Ryan, D.G.; Prag, H.A.; Dikovskaya, D.; Menon, D.; Zaslona, Z.; Jedrychowski, M.P.; Costa, A.S.H.; Higgins, M.; Hams, E.; et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 2018, 556, 113–117.
- Eggler, A.L.; Liu, G.; Pezzuto, J.M.; van Breemen, R.B.; Mesecar, A.D. Modifying specific cysteines of the electrophile-sensing human Keap1 protein is insufficient to disrupt binding to the Nrf2 domain Neh2. Proc. Natl. Acad. Sci. USA 2005, 102, 10070–10075.
- Shin, J.W.; Chun, K.S.; Kim, D.H.; Kim, S.J.; Kim, S.H.; Cho, N.C.; Na, H.K.; Surh, Y.J. Curcumin induces stabilization of Nrf2 protein through Keap1 cysteine modification. Biochem. Pharmacol. 2020, 173, 113820.
- Kerr, F.; Sofola-Adesakin, O.; Ivanov, D.K.; Gatliff, J.; Gomez Perez-Nievas, B.; Bertrand, H.C.; Martinez, P.; Callard, R.; Snoeren, I.; Cocheme, H.M.; et al. Direct Keap1-Nrf2 disruption as a potential therapeutic target for Alzheimer’s disease. PLoS Genet. 2017, 13, e1006593.
- Chowdhry, S.; Zhang, Y.; McMahon, M.; Sutherland, C.; Cuadrado, A.; Hayes, J.D. Nrf2 is controlled by two distinct beta-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 2013, 32, 3765–3781.
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223.
- Covas, G.; Marinho, H.S.; Cyrne, L.; Antunes, F. Activation of Nrf2 by H2O2: De novo synthesis versus nuclear translocation. Methods Enzymol. 2013, 528, 157–171.
- Casares, L.; Unciti-Broceta, J.D.; Prados, M.E.; Caprioglio, D.; Mattoteia, D.; Higgins, M.; Apendino, G.; Dinkova-Kostova, A.T.; Munoz, E.; de la Vega, L. Isomeric O-methyl cannabidiolquinones with dual BACH1/NRF2 activity. Redox Biol. 2020, 37, 101689.
- Mrowietz, U.; Morrison, P.J.; Suhrkamp, I.; Kumanova, M.; Clement, B. The Pharmacokinetics of Fumaric Acid Esters Reveal Their In Vivo Effects. Trends Pharmacol. Sci. 2018, 39, 1–12.
- Belcher, J.D.; Chen, C.; Nguyen, J.; Zhang, P.; Abdulla, F.; Nguyen, P.; Killeen, T.; Xu, P.; O’Sullivan, G.; Nath, K.A.; et al. Control of Oxidative Stress and Inflammation in Sickle Cell Disease with the Nrf2 Activator Dimethyl Fumarate. Antioxid. Redox Signal. 2017, 26, 748–762.
- Ahuja, M.; Ammal Kaidery, N.; Yang, L.; Calingasan, N.; Smirnova, N.; Gaisin, A.; Gaisina, I.N.; Gazaryan, I.; Hushpulian, D.M.; Kaddour-Djebbar, I.; et al. Distinct Nrf2 Signaling Mechanisms of Fumaric Acid Esters and Their Role in Neuroprotection against 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine-Induced Experimental Parkinson’s-Like Disease. J. Neurosci. 2016, 36, 6332–6351.
- Galloway, D.A.; Williams, J.B.; Moore, C.S. Effects of fumarates on inflammatory human astrocyte responses and oligodendrocyte differentiation. Ann. Clin. Transl. Neurol. 2017, 4, 381–391.
- Higashi, C.; Kawaji, A.; Tsuda, N.; Hayashi, M.; Saito, R.; Yagishita, Y.; Suzuki, T.; Uruno, A.; Nakamura, M.; Nakao, K.; et al. The novel Nrf2 inducer TFM-735 ameliorates experimental autoimmune encephalomyelitis in mice. Eur. J. Pharmacol. 2017, 802, 76–84.
- Delmastro-Greenwood, M.; Hughan, K.S.; Vitturi, D.A.; Salvatore, S.R.; Grimes, G.; Potti, G.; Shiva, S.; Schopfer, F.J.; Gladwin, M.T.; Freeman, B.A.; et al. Nitrite and nitrate-dependent generation of anti-inflammatory fatty acid nitroalkenes. Free Radic. Biol. Med. 2015, 89, 333–341.
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, S.; Levonen, A.L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317.
- Lu, M.C.; Zhao, J.; Liu, Y.T.; Liu, T.; Tao, M.M.; You, Q.D.; Jiang, Z.Y. CPUY192018, a potent inhibitor of the Keap1-Nrf2 protein-protein interaction, alleviates renal inflammation in mice by restricting oxidative stress and NF-kappaB activation. Redox Biol. 2019, 26, 101266.
- Moi, P.; Chan, K.; Asunis, I.; Cao, A.; Kan, Y.W. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc. Natl. Acad. Sci. USA 1994, 91, 9926–9930.
- Lu, M.C.; Ji, J.A.; Jiang, Y.L.; Chen, Z.Y.; Yuan, Z.W.; You, Q.D.; Jiang, Z.Y. An inhibitor of the Keap1-Nrf2 protein-protein interaction protects NCM460 colonic cells and alleviates experimental colitis. Sci. Rep. 2016, 6, 26585.
- Hu, L.; Magesh, S.; Chen, L.; Wang, L.; Lewis, T.A.; Chen, Y.; Khodier, C.; Inoyama, D.; Beamer, L.J.; Emge, T.J.; et al. Discovery of a small-molecule inhibitor and cellular probe of Keap1-Nrf2 protein-protein interaction. Bioorg. Med. Chem. Lett. 2013, 23, 3039–3043.
- Marcotte, D.; Zeng, W.; Hus, J.C.; McKenzie, A.; Hession, C.; Jin, P.; Bergeron, C.; Lugovskoy, A.; Enyedy, I.; Cuervo, H.; et al. Small molecules inhibit the interaction of Nrf2 and the Keap1 Kelch domain through a non-covalent mechanism. Bioorg. Med. Chem. 2013, 21, 4011–4019.
- Lignitto, L.; LeBoeuf, S.E.; Homer, H.; Jiang, S.; Askenazi, M.; Karakousi, T.R.; Pass, H.I.; Bhutkar, A.J.; Tsirigos, A.; Ueberheide, B.; et al. Nrf2 Activation Promotes Lung Cancer Metastasis by Inhibiting the Degradation of Bach1. Cell 2019, 178, 316–329e.318.
- Tossetta, G.; Fantone, S.; Montanari, E.; Marzioni, D.; Goteri, G. Role of NRF2 in Ovarian Cancer. Antioxidants 2022, 11, 663.
- Barrera, G.; Cucci, M.A.; Grattarola, M.; Dianzani, C.; Muzio, G.; Pizzimenti, S. Control of Oxidative Stress in Cancer Chemoresistance: Spotlight on Nrf2 Role. Antioxidants 2021, 10, 510.
- Pedre, B.; Barayeu, U.; Ezerina, D.; Dick, T.P. The mechanism of action of N-acetylcysteine (NAC): The emerging role of H2S and sulfane sulfur species. Pharmacol. Ther. 2021, 228, 107916.
- Pilewskie, M.; Morrow, M. Axillary Nodal Management Following Neoadjuvant Chemotherapy: A Review. JAMA Oncol. 2017, 3, 549–555.
- Piskounova, E.; Agathocleous, M.; Murphy, M.M.; Hu, Z.; Huddlestun, S.E.; Zhao, Z.; Leitch, A.M.; Johnson, T.M.; DeBerardinis, R.J.; Morrison, S.J. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 2015, 527, 186–191.
- Cacciatore, I.; Cornacchia, C.; Pinnen, F.; Mollica, A.; Di Stefano, A. Prodrug approach for increasing cellular glutathione levels. Molecules 2010, 15, 1242–1264.
- Puri, R.N.; Meister, A. Transport of glutathione, as gamma-glutamylcysteinylglycyl ester, into liver and kidney. Proc. Natl. Acad. Sci. USA 1983, 80, 5258–5260.
- Gomes-Neto, J.C.; Round, J.L. Gut microbiota: A new way to take your vitamins. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 521–522.
- Said, H.M.; Nexo, E. Gastrointestinal Handling of Water-Soluble Vitamins. Compr. Physiol. 2018, 8, 1291–1311.
- Cossey, L.N.; Rahim, F.; Larsen, C.P. Oxalate nephropathy and intravenous vitamin C. Am. J. Kidney Dis. 2013, 61, 1032–1035.
- Ngo, B.; Van Riper, J.M.; Cantley, L.C.; Yun, J. Targeting cancer vulnerabilities with high-dose vitamin C. Nat. Rev. Cancer 2019, 19, 271–282.
- Mikirova, N.; Casciari, J.; Rogers, A.; Taylor, P. Effect of high-dose intravenous vitamin C on inflammation in cancer patients. J. Transl. Med. 2012, 10, 189.
- Yun, J.; Mullarky, E.; Lu, C.; Bosch, K.N.; Kavalier, A.; Rivera, K.; Roper, J.; Chio, I.I.; Giannopoulou, E.G.; Rago, C.; et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 2015, 350, 1391–1396.
- Harris, H.R.; Orsini, N.; Wolk, A. Vitamin C and survival among women with breast cancer: A meta-analysis. Eur. J. Cancer 2014, 50, 1223–1231.
- Carr, A.C.; Cook, J. Intravenous Vitamin C for Cancer Therapy—Identifying the Current Gaps in Our Knowledge. Front. Physiol. 2018, 9, 1182.
- Creagan, E.T.; Moertel, C.G.; O’Fallon, J.R.; Schutt, A.J.; O’Connell, M.J.; Rubin, J.; Frytak, S. Failure of high-dose vitamin C (ascorbic acid) therapy to benefit patients with advanced cancer. A controlled trial. N. Engl. J. Med. 1979, 301, 687–690.
- Moertel, C.G.; Fleming, T.R.; Creagan, E.T.; Rubin, J.; O’Connell, M.J.; Ames, M.M. High-dose vitamin C versus placebo in the treatment of patients with advanced cancer who have had no prior chemotherapy. A randomized double-blind comparison. N. Engl. J. Med. 1985, 312, 137–141.
- Niki, E. Role of vitamin E as a lipid-soluble peroxyl radical scavenger: In vitro and in vivo evidence. Free Radic. Biol. Med. 2014, 66, 3–12.
- Sauberlich, H.E. Pharmacology of vitamin C. Annu. Rev. Nutr. 1994, 14, 371–391.
- Yang, C.S.; Suh, N.; Kong, A.N. Does vitamin E prevent or promote cancer? Cancer Prev. Res. 2012, 5, 701–705.
- Konate, M.M.; Antony, S.; Doroshow, J.H. Inhibiting the Activity of NADPH Oxidase in Cancer. Antioxid. Redox Signal. 2020, 33, 435–454.
- Cifuentes-Pagano, M.E.; Meijles, D.N.; Pagano, P.J. Nox Inhibitors & Therapies: Rational Design of Peptidic and Small Molecule Inhibitors. Curr. Pharm. Des. 2015, 21, 6023–6035.
- Cifuentes-Pagano, E.; Csanyi, G.; Pagano, P.J. NADPH oxidase inhibitors: A decade of discovery from Nox2ds to HTS. Cell Mol. Life Sci. 2012, 69, 2315–2325.
- Altenhofer, S.; Radermacher, K.A.; Kleikers, P.W.; Wingler, K.; Schmidt, H.H. Evolution of NADPH Oxidase Inhibitors: Selectivity and Mechanisms for Target Engagement. Antioxid. Redox Signal. 2015, 23, 406–427.
- DiStasi, M.R.; Mund, J.A.; Bohlen, H.G.; Miller, S.J.; Ingram, D.A.; Dalsing, M.C.; Unthank, J.L. Impaired compensation to femoral artery ligation in diet-induced obese mice is primarily mediated via suppression of collateral growth by Nox2 and p47phox. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1207–H1217.
- Krotz, F.; Sohn, H.Y.; Gloe, T.; Zahler, S.; Riexinger, T.; Schiele, T.M.; Becker, B.F.; Theisen, K.; Klauss, V.; Pohl, U. NAD(P)H oxidase-dependent platelet superoxide anion release increases platelet recruitment. Blood 2002, 100, 917–924.
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313.
- Ebrahimian, T.; Li, M.W.; Lemarie, C.A.; Simeone, S.M.; Pagano, P.J.; Gaestel, M.; Paradis, P.; Wassmann, S.; Schiffrin, E.L. Mitogen-activated protein kinase-activated protein kinase 2 in angiotensin II-induced inflammation and hypertension: Regulation of oxidative stress. Hypertension 2011, 57, 245–254.
- Hou, L.; Zhang, L.; Hong, J.S.; Zhang, D.; Zhao, J.; Wang, Q. Nicotinamide Adenine Dinucleotide Phosphate Oxidase and Neurodegenerative Diseases: Mechanisms and Therapy. Antioxid. Redox Signal. 2020, 33, 374–393.
- Doroshow, J.H.; Gaur, S.; Markel, S.; Lu, J.; van Balgooy, J.; Synold, T.W.; Xi, B.; Wu, X.; Juhasz, A. Effects of iodonium-class flavin dehydrogenase inhibitors on growth, reactive oxygen production, cell cycle progression, NADPH oxidase 1 levels, and gene expression in human colon cancer cells and xenografts. Free Radic. Biol. Med. 2013, 57, 162–175.
- Schroder, K. NADPH oxidases: Current aspects and tools. Redox Biol. 2020, 34, 101512.
- Mora-Pale, M.; Kwon, S.J.; Linhardt, R.J.; Dordick, J.S. Trimer hydroxylated quinone derived from apocynin targets cysteine residues of p47phox preventing the activation of human vascular NADPH oxidase. Free Radic. Biol. Med. 2012, 52, 962–969.
- Murley, J.S.; Arbiser, J.L.; Weichselbaum, R.R.; Grdina, D.J. ROS modifiers and NOX4 affect the expression of the survivin-associated radio-adaptive response. Free Radic. Biol. Med. 2018, 123, 39–52.
- Bhandarkar, S.S.; Jaconi, M.; Fried, L.E.; Bonner, M.Y.; Lefkove, B.; Govindarajan, B.; Perry, B.N.; Parhar, R.; Mackelfresh, J.; Sohn, A.; et al. Fulvene-5 potently inhibits NADPH oxidase 4 and blocks the growth of endothelial tumors in mice. J. Clin. Investig. 2009, 119, 2359–2365.
- Cao, F.; Zhang, L.; You, Y.; Zheng, L.; Ren, J.; Qu, X. An Enzyme-Mimicking Single-Atom Catalyst as an Efficient Multiple Reactive Oxygen and Nitrogen Species Scavenger for Sepsis Management. Angew. Chem. Int. Ed. Engl. 2020, 59, 5108–5115.
- Carillon, J.; Rouanet, J.M.; Cristol, J.P.; Brion, R. Superoxide dismutase administration, a potential therapy against oxidative stress related diseases: Several routes of supplementation and proposal of an original mechanism of action. Pharm. Res. 2013, 30, 2718–2728.
- Johnke, R.M.; Sattler, J.A.; Allison, R.R. Radioprotective agents for radiation therapy: Future trends. Future Oncol. 2014, 10, 2345–2357.
- Weitner, T.; Kos, I.; Sheng, H.; Tovmasyan, A.; Reboucas, J.S.; Fan, P.; Warner, D.S.; Vujaskovic, Z.; Batinic-Haberle, I.; Spasojevic, I. Comprehensive pharmacokinetic studies and oral bioavailability of two Mn porphyrin-based SOD mimics, MnTE-2-PyP5+ and MnTnHex-2-PyP5+. Free Radic. Biol. Med. 2013, 58, 73–80.
- Tovmasyan, A.; Reboucas, J.S.; Benov, L. Simple biological systems for assessing the activity of superoxide dismutase mimics. Antioxid. Redox Signal. 2014, 20, 2416–2436.
- Bonetta, R. Potential Therapeutic Applications of MnSODs and SOD-Mimetics. Chemistry 2018, 24, 5032–5041.
- Sherlock, L.G.; Trumpie, A.; Hernandez-Lagunas, L.; McKenna, S.; Fisher, S.; Bowler, R.; Wright, C.J.; Delaney, C.; Nozik-Grayck, E. Redistribution of Extracellular Superoxide Dismutase Causes Neonatal Pulmonary Vascular Remodeling and PH but Protects Against Experimental Bronchopulmonary Dysplasia. Antioxidants 2018, 7, 42.
- Chatterjee, A.; Zhu, Y.; Tong, Q.; Kosmacek, E.A.; Lichter, E.Z.; Oberley-Deegan, R.E. The Addition of Manganese Porphyrins during Radiation Inhibits Prostate Cancer Growth and Simultaneously Protects Normal Prostate Tissue from Radiation Damage. Antioxidants 2018, 7, 21.
- Batinic-Haberle, I.; Tovmasyan, A.; Roberts, E.R.; Vujaskovic, Z.; Leong, K.W.; Spasojevic, I. SOD therapeutics: Latest insights into their structure-activity relationships and impact on the cellular redox-based signaling pathways. Antioxid. Redox Signal. 2014, 20, 2372–2415.
- Batinic-Haberle, I.; Tome, M.E. Thiol regulation by Mn porphyrins, commonly known as SOD mimics. Redox Biol. 2019, 25, 101139.
- Sishc, B.J.; Ding, L.; Nam, T.K.; Heer, C.D.; Rodman, S.N.; Schoenfeld, J.D.; Fath, M.A.; Saha, D.; Pulliam, C.F.; Langen, B.; et al. Avasopasem manganese synergizes with hypofractionated radiation to ablate tumors through the generation of hydrogen peroxide. Sci. Transl. Med. 2021, 13, eabb3768.
- Heer, C.D.; Davis, A.B.; Riffe, D.B.; Wagner, B.A.; Falls, K.C.; Allen, B.G.; Buettner, G.R.; Beardsley, R.A.; Riley, D.P.; Spitz, D.R. Superoxide Dismutase Mimetic GC4419 Enhances the Oxidation of Pharmacological Ascorbate and Its Anticancer Effects in an H(2)O(2)-Dependent Manner. Antioxidants 2018, 7, 18.
- El-Mahdy, M.A.; Alzarie, Y.A.; Hemann, C.; Badary, O.A.; Nofal, S.; Zweier, J.L. The novel SOD mimetic GC4419 increases cancer cell killing with sensitization to ionizing radiation while protecting normal cells. Free Radic. Biol. Med. 2020, 160, 630–642.
- Asadi, Z.; Mandegani, Z.; Asadi, M.; Pakiari, A.H.; Salarhaji, M.; Manassir, M.; Karbalaei-Heidari, H.R.; Rastegari, B.; Sedaghat, M. Substituted effect on some water-soluble Mn(II) salen complexes: DNA binding, cytotoxicity, molecular docking, DFT studies and theoretical IR & UV studies. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2019, 206, 278–294.
More