Staphylococcus aureus is a very common Gram-positive bacterium, and S. aureus infections play an extremely important role in a variety of diseases. Staphylococcus aureus (S. aureus), a Gram-positive bacterium, is one of the most notorious human pathogens, causing illnesses ranging from mild skin and wound infections to fatal sepsis or multi-organ failure. Inflammatory cells play an important role in S. aureus infection. S. aureus infection and toxins can activate a variety of inflammatory cells, such as keratinocytes, helper T cells, innate lymphoid cells (ILCs), macrophages, dendritic cells (DCs), mast cells, neutrophils, eosinophils, and basophils, which release inflammatory factors that accumulate at the site of infection and cause an inflammatory response.
- Staphylococcus aureus
- toxin
- inflammatory cells
- pyroptosis
1. Virulence Factors of S. aureus
1.1. Secreted Virulence Factors
1.1.1. PFTs
-
Hla, Hlb, and Hlg
- 2.
-
α-Toxin
- 3.
-
PVL
1.1.2. PSMs
-
PSMα
- 2.
-
PSMβ
- 3.
-
PSMγ (δ Toxin)
1.1.3. Proteases
-
ETs
- 2.
-
Serine Protease-Like Proteins (Spls)
- 3.
-
Staphopain B (SspB)
1.1.4. SAgs
-
SEs
| SEs | Functions |
|---|---|
| SEA | Anti-protein hydrolase, supercarcinogenic and emetic toxin, causes food poisoning, but no diarrheal activity |
| SEB | Increase in the number of T lymphocytes, as well as the concentration of pro-inflammatory cytokines and inflammatory mediators in the intestinal mucosa; increase mucosal permeability and intestinal secretion |
| SEC | Supercarcinogenic and emetic toxin, but no diarrheal activity |
| SEG | Reversible destruction of intestinal cell ultrastructure |
| SEH | Causes food poisoning |
| SEI | Reversibly disrupts enterocyte ultrastructure and exhibits weak emetic activity |
| SEM | Strong emetic potential |
| SEO | Has emetic activity |
| SEQ | Significantly stable for heat treatment and digestive enzyme degradation, and exhibits significant supercarcinogenic and emetic activity |
- 2.
-
TSST-1
1.1.5. Secreted Enzymes (Exoenzymes) and Effectors
-
EsxA and EsxB
- 2.
-
Coa
- 3.
-
Nuc and AdsA
- 4.
-
Extracellular Adhesion Protein (Eap)
1.2. Surface Proteins of S. aureus (Cell Wall-Anchored (CWA) Proteins)
1.2.1. MSCRAMM
1.2.2. Staphylococcal Protein A (SpA)
1.3. PAMPs
1.3.1. Triacyl Lipopetides and Diacyl Lipoproteins
1.3.2. LTA
1.3.3. PGN
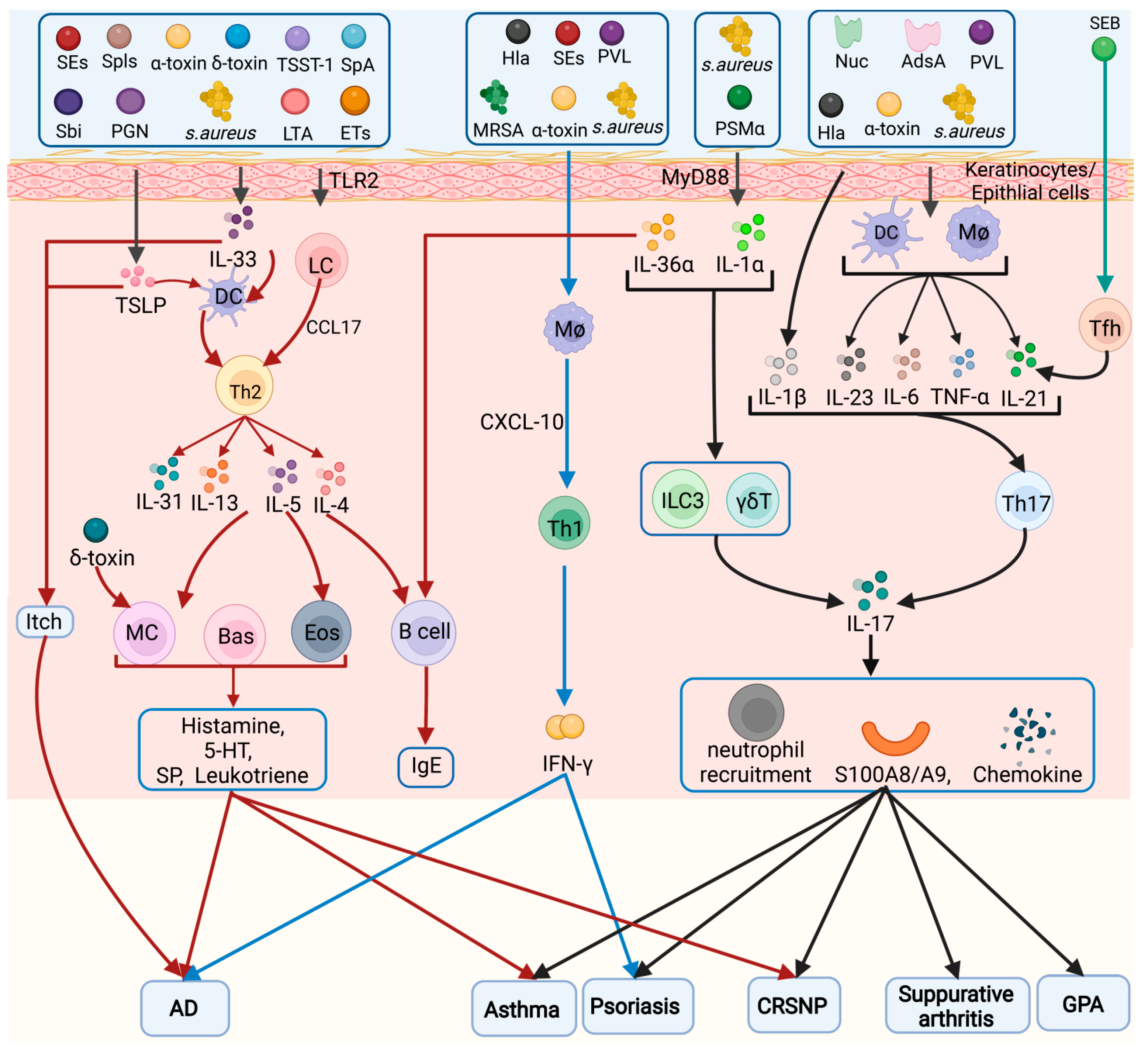
2. Different Inflammatory Cell Types Involved in
Pathogenesis
2.1. Keratinocytes/Epithelial Cells
2.2. Helper T Cells (Th Cells)
2.2.1. T Helper 1 (Th1) Cells
2.2.2. Th2 Cells
2.2.3. Th17 Cells
2.2.4. Tregs
2.2.5. Tfh Cells
2.2.6. Th9 Cells
2.3. ILCs
2.4. Macrophages
2.5. DCs
2.6. Mast Cells
2.7. Neutrophils
2.8. Eosinophils (Eos)
2.9. Basophils
2.10. B Cells
References
- Tam, K.; Torres, V.J. Staphylococcus aureus Secreted Toxins and Extracellular Enzymes. Microbiol. Spectr. 2019, 7, 2.
- Foster, T.J.; Geoghegan, J.A.; Ganesh, V.K.; Höök, M. Adhesion, invasion and evasion: The many functions of the surface proteins of Staphylococcus aureus. Nat. Rev. Microbiol. 2014, 12, 49–62.
- Askarian, F.; Wagner, T.; Johannessen, M.; Nizet, V. Staphylococcus aureus modulation of innate immune responses through Toll-like (TLR), (NOD)-like (NLR) and C-type lectin (CLR) receptors. FEMS Microbiol. Rev. 2018, 42, 656–671.
- Verma, P.; Gandhi, S.; Lata, K.; Chattopadhyay, K. Pore-forming toxins in infection and immunity. Biochem. Soc. Trans. 2021, 49, 455–465.
- Otto, M. Phenol-soluble modulins. Int. J. Med. Microbiol. 2014, 304, 164–169.
- Yagi, S.; Wakaki, N.; Ikeda, N.; Takagi, Y.; Uchida, H.; Kato, Y.; Minamino, M. Presence of staphylococcal exfoliative toxin A in sera of patients with atopic dermatitis. Clin. Exp. Allergy 2004, 34, 984–993.
- Spaulding, A.R.; Salgado-Pabón, W.; Kohler, P.L.; Horswill, A.R.; Leung, D.Y.; Schlievert, P.M. Staphylococcal and streptococcal superantigen exotoxins. Clin. Microbiol. Rev. 2013, 26, 422–447.
- Cassidy, P.; Harshman, S. Studies on the binding of staphylococcal 125I-labeled alpha-toxin to rabbit erythrocytes. Biochemistry 1976, 15, 2348–2355.
- Becker, K.A.; Fahsel, B.; Kemper, H.; Mayeres, J.; Li, C.; Wilker, B.; Keitsch, S.; Soddemann, M.; Sehl, C.; Kohnen, M.; et al. Staphylococcus aureus Alpha-Toxin Disrupts Endothelial-Cell Tight Junctions via Acid Sphingomyelinase and Ceramide. Infect. Immun. 2018, 86, e00606-17.
- Muñoz-Planillo, R.; Franchi, L.; Miller, L.S.; Núñez, G. A critical role for hemolysins and bacterial lipoproteins in Staphylococcus aureus-induced activation of the Nlrp3 inflammasome. J. Immunol. 2009, 183, 3942–3948.
- Mulcahy, M.E.; O’Brien, E.C.; O’Keeffe, K.M.; Vozza, E.G.; Leddy, N.; McLoughlin, R.M. Manipulation of Autophagy and Apoptosis Facilitates Intracellular Survival of Staphylococcus aureus in Human Neutrophils. Front. Immunol. 2020, 11, 565545.
- Adler, A.; Temper, V.; Block, C.S.; Abramson, N.; Moses, A.E. Panton-Valentine leukocidin-producing Staphylococcus aureus. Emerg. Infect. Dis. 2006, 12, 1789–1790.
- Wiseman, G.M. The hemolysins of Staphylococcus aureus. Bacteriol. Rev. 1975, 39, 317–344.
- Bubeck Wardenburg, J.; Patel, R.J.; Schneewind, O. Surface proteins and exotoxins are required for the pathogenesis of Staphylococcus aureus pneumonia. Infect. Immun. 2007, 75, 1040–1044.
- Lizak, M.; Yarovinsky, T.O. Phospholipid scramblase 1 mediates type i interferon-induced protection against staphylococcal α-toxin. Cell Host Microbe 2012, 11, 70–80.
- Bhakdi, S.; Muhly, M.; Korom, S.; Hugo, F. Release of interleukin-1 beta associated with potent cytocidal action of staphylococcal alpha-toxin on human monocytes. Infect. Immun. 1989, 57, 3512–3519.
- Suttorp, N.; Fuhrmann, M.; Tannert-Otto, S.; Grimminger, F.; Bhadki, S. Pore-forming bacterial toxins potently induce release of nitric oxide in porcine endothelial cells. J. Exp. Med. 1993, 178, 337–341.
- Craven, R.R.; Gao, X.; Allen, I.C.; Gris, D.; Bubeck Wardenburg, J.; McElvania-Tekippe, E.; Ting, J.P.; Duncan, J.A. Staphylococcus aureus alpha-hemolysin activates the NLRP3-inflammasome in human and mouse monocytic cells. PLoS ONE 2009, 4, e7446.
- Suttorp, N.; Seeger, W.; Dewein, E.; Bhakdi, S.; Roka, L. Staphylococcal alpha-toxin-induced PGI2 production in endothelial cells: Role of calcium. Am. J. Physiol. 1985, 248, C127–C134.
- Berube, B.J.; Bubeck Wardenburg, J. Staphylococcus aureus α-toxin: Nearly a century of intrigue. Toxins 2013, 5, 1140–1166.
- Grimminger, F.; Rose, F.; Sibelius, U.; Meinhardt, M.; Pötzsch, B.; Spriestersbach, R.; Bhakdi, S.; Suttorp, N.; Seeger, W. Human endothelial cell activation and mediator release in response to the bacterial exotoxins Escherichia coli hemolysin and staphylococcal alpha-toxin. J. Immunol. 1997, 159, 1909–1916.
- Iacovache, I.; Bischofberger, M.; van der Goot, F.G. Structure and assembly of pore-forming proteins. Curr. Opin. Struct. Biol. 2010, 20, 241–246.
- Breuer, K.; Wittmann, M.; Kempe, K.; Kapp, A.; Mai, U.; Dittrich-Breiholz, O.; Kracht, M.; Mrabet-Dahbi, S.; Werfel, T. Alpha-toxin is produced by skin colonizing Staphylococcus aureus and induces a T helper type 1 response in atopic dermatitis. Clin. Exp. Allergy 2005, 35, 1088–1095.
- Kasraie, S.; Niebuhr, M.; Kopfnagel, V.; Dittrich-Breiholz, O.; Kracht, M.; Werfel, T. Macrophages from patients with atopic dermatitis show a reduced CXCL10 expression in response to staphylococcal α-toxin. Allergy 2012, 67, 41–49.
- Schlievert, P.M.; Roller, R.J.; Kilgore, S.H.; Villarreal, M.; Klingelhutz, A.J.; Leung, D.Y.M. Staphylococcal TSST-1 Association with Eczema Herpeticum in Humans. mSphere 2021, 6, e00608-21.
- Okano, M.; Fujiwara, T.; Kariya, S.; Higaki, T.; Haruna, T.; Matsushita, O.; Noda, Y.; Makihara, S.; Kanai, K.; Noyama, Y.; et al. Cellular responses to Staphylococcus aureus alpha-toxin in chronic rhinosinusitis with nasal polyps. Allergol. Int. 2014, 63, 563–573.
- Kang, S.S.; Noh, S.Y.; Park, O.J.; Yun, C.H.; Han, S.H. Staphylococcus aureus induces IL-8 expression through its lipoproteins in the human intestinal epithelial cell, Caco-2. Cytokine 2015, 75, 174–180.
- Kwak, Y.K.; Vikström, E.; Magnusson, K.E.; Vécsey-Semjén, B.; Colque-Navarro, P.; Möllby, R. The Staphylococcus aureus alpha-toxin perturbs the barrier function in Caco-2 epithelial cell monolayers by altering junctional integrity. Infect. Immun. 2012, 80, 1670–1680.
- Gillet, Y.; Issartel, B.; Vanhems, P.; Fournet, J.C.; Lina, G.; Bes, M.; Vandenesch, F.; Piémont, Y.; Brousse, N.; Floret, D.; et al. Association between Staphylococcus aureus strains carrying gene for Panton-Valentine leukocidin and highly lethal necrotising pneumonia in young immunocompetent patients. Lancet 2002, 359, 753–759.
- Finck-Barbançon, V.; Duportail, G.; Meunier, O.; Colin, D.A. Pore formation by a two-component leukocidin from Staphylococcus aureus within the membrane of human polymorphonuclear leukocytes. Biochim. Biophys. Acta 1993, 1182, 275–282.
- Yanai, M.; Rocha, M.A.; Matolek, A.Z.; Chintalacharuvu, A.; Taira, Y.; Chintalacharuvu, K.; Beenhouwer, D.O. Separately or combined, LukG/LukH is functionally unique compared to other staphylococcal bicomponent leukotoxins. PLoS ONE 2014, 9, e89308.
- Melehani, J.H.; James, D.B.; DuMont, A.L.; Torres, V.J.; Duncan, J.A. Staphylococcus aureus Leukocidin A/B (LukAB) Kills Human Monocytes via Host NLRP3 and ASC when Extracellular, but Not Intracellular. PLoS Pathog. 2015, 11, e1004970.
- Holzinger, D.; Gieldon, L.; Mysore, V.; Nippe, N.; Taxman, D.J.; Duncan, J.A.; Broglie, P.M.; Marketon, K.; Austermann, J.; Vogl, T.; et al. Staphylococcus aureus Panton-Valentine leukocidin induces an inflammatory response in human phagocytes via the NLRP3 inflammasome. J. Leukoc. Biol. 2012, 92, 1069–1081.
- Spaan, A.N.; Vrieling, M.; Wallet, P.; Badiou, C.; Reyes-Robles, T.; Ohneck, E.A.; Benito, Y.; de Haas, C.J.; Day, C.J.; Jennings, M.P.; et al. The staphylococcal toxins γ-haemolysin AB and CB differentially target phagocytes by employing specific chemokine receptors. Nat. Commun. 2014, 5, 5438.
- Staali, L.; Monteil, H.; Colin, D.A. The staphylococcal pore-forming leukotoxins open Ca2+ channels in the membrane of human polymorphonuclear neutrophils. J. Membr. Biol. 1998, 162, 209–216.
- Noda, M.; Kato, I.; Hirayama, T.; Matsuda, F. Mode of action of staphylococcal leukocidin: Effects of the S and F components on the activities of membrane-associated enzymes of rabbit polymorphonuclear leukocytes. Infect. Immun. 1982, 35, 38–45.
- Chow, S.H.; Deo, P.; Yeung, A.T.Y.; Kostoulias, X.P.; Jeon, Y.; Gao, M.L.; Seidi, A.; Olivier, F.A.B.; Sridhar, S.; Nethercott, C.; et al. Targeting NLRP3 and Staphylococcal pore-forming toxin receptors in human-induced pluripotent stem cell-derived macrophages. J. Leukoc. Biol. 2020, 108, 967–981.
- Kitur, K.; Parker, D.; Nieto, P.; Ahn, D.S.; Cohen, T.S.; Chung, S.; Wachtel, S.; Bueno, S.; Prince, A. Toxin-induced necroptosis is a major mechanism of Staphylococcus aureus lung damage. PLoS Pathog. 2015, 11, e1004820.
- Shallcross, L.J.; Fragaszy, E.; Johnson, A.M.; Hayward, A.C. The role of the Panton-Valentine leucocidin toxin in staphylococcal disease: A systematic review and meta-analysis. Lancet Infect. Dis. 2013, 13, 43–54.
- Genestier, A.L.; Michallet, M.C.; Prévost, G.; Bellot, G.; Chalabreysse, L.; Peyrol, S.; Thivolet, F.; Etienne, J.; Lina, G.; Vallette, F.M.; et al. Staphylococcus aureus Panton-Valentine leukocidin directly targets mitochondria and induces Bax-independent apoptosis of human neutrophils. J. Clin. Investig. 2005, 115, 3117–3127.
- Huang, J.; Zhang, T.; Zou, X.; Wu, S.; Zhu, J. Panton-valentine leucocidin carrying Staphylococcus aureus causing necrotizing pneumonia inactivates the JAK/STAT signaling pathway and increases the expression of inflammatory cytokines. Infect. Genet. Evol. 2020, 86, 104582.
- Nakagawa, S.; Matsumoto, M.; Katayama, Y.; Oguma, R.; Wakabayashi, S.; Nygaard, T.; Saijo, S.; Inohara, N.; Otto, M.; Matsue, H.; et al. Staphylococcus aureus Virulent PSMα Peptides Induce Keratinocyte Alarmin Release to Orchestrate IL-17-Dependent Skin Inflammation. Cell Host Microbe 2017, 22, 667–677.e5.
- Gray, B.; Hall, P.; Gresham, H. Targeting agr- and agr-Like quorum sensing systems for development of common therapeutics to treat multiple gram-positive bacterial infections. Sensors 2013, 13, 5130–5166.
- Zhou, Y.; Niu, C.; Ma, B.; Xue, X.; Li, Z.; Chen, Z.; Li, F.; Zhou, S.; Luo, X.; Hou, Z. Inhibiting PSMα-induced neutrophil necroptosis protects mice with MRSA pneumonia by blocking the agr system. Cell Death Dis. 2018, 9, 362.
- Hanzelmann, D.; Joo, H.S.; Franz-Wachtel, M.; Hertlein, T.; Stevanovic, S.; Macek, B.; Wolz, C.; Götz, F.; Otto, M.; Kretschmer, D.; et al. Toll-like receptor 2 activation depends on lipopeptide shedding by bacterial surfactants. Nat. Commun. 2016, 7, 12304.
- Patrick, G.J.; Liu, H.; Alphonse, M.P.; Dikeman, D.A.; Youn, C.; Otterson, J.C.; Wang, Y.; Ravipati, A.; Mazhar, M.; Denny, G.; et al. Epicutaneous Staphylococcus aureus induces IL-36 to enhance IgE production and ensuing allergic disease. J. Clin. Investig. 2021, 131, e143334.
- McKevitt, A.I.; Bjornson, G.L.; Mauracher, C.A.; Scheifele, D.W. Amino acid sequence of a deltalike toxin from Staphylococcus epidermidis. Infect. Immun. 1990, 58, 1473–1475.
- Kretschmer, D.; Gleske, A.K.; Rautenberg, M.; Wang, R.; Köberle, M.; Bohn, E.; Schöneberg, T.; Rabiet, M.J.; Boulay, F.; Klebanoff, S.J.; et al. Human formyl peptide receptor 2 senses highly pathogenic Staphylococcus aureus. Cell Host Microbe 2010, 7, 463–473.
- Rautenberg, M.; Joo, H.S.; Otto, M.; Peschel, A. Neutrophil responses to staphylococcal pathogens and commensals via the formyl peptide receptor 2 relates to phenol-soluble modulin release and virulence. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2011, 25, 1254–1263.
- Nakamura, Y.; Oscherwitz, J.; Cease, K.B.; Chan, S.M.; Muñoz-Planillo, R.; Hasegawa, M.; Villaruz, A.E.; Cheung, G.Y.; McGavin, M.J.; Travers, J.B.; et al. Staphylococcus δ-toxin induces allergic skin disease by activating mast cells. Nature 2013, 503, 397–401.
- Kapral, F.A. Staphylococcus aureus delta toxin as an enterotoxin. Ciba. Found. Symp. 1985, 112, 215–229.
- Ullah, A.; Khan, A.; Al-Harrasi, A.; Ullah, K.; Shabbir, A. Three-Dimensional Structure Characterization and Inhibition Study of Exfoliative Toxin D from Staphylococcus aureus. Front. Pharmacol. 2022, 13, 800970.
- Imanishi, I.; Nicolas, A.; Caetano, A.B.; Castro, T.L.P.; Tartaglia, N.R.; Mariutti, R.; Guédon, E.; Even, S.; Berkova, N.; Arni, R.K.; et al. Exfoliative toxin E, a new Staphylococcus aureus virulence factor with host-specific activity. Sci. Rep. 2019, 9, 16336.
- Stentzel, S.; Teufelberger, A.; Nordengrün, M.; Kolata, J.; Schmidt, F.; van Crombruggen, K.; Michalik, S.; Kumpfmüller, J.; Tischer, S.; Schweder, T.; et al. Staphylococcal serine protease-like proteins are pacemakers of allergic airway reactions to Staphylococcus aureus. J. Allergy Clin. Immunol. 2017, 139, 492–500.e8.
- Melish, M.E.; Glasgow, L.A. Staphylococcal scalded skin syndrome: The expanded clinical syndrome. J. Pediatr. 1971, 78, 958–967.
- Bukowski, M.; Wladyka, B.; Dubin, G. Exfoliative toxins of Staphylococcus aureus. Toxins 2010, 2, 1148–1165.
- Nishifuji, K.; Sugai, M.; Amagai, M. Staphylococcal exfoliative toxins: “molecular scissors” of bacteria that attack the cutaneous defense barrier in mammals. J. Dermatol. Sci. 2008, 49, 21–31.
- Amagai, M.; Yamaguchi, T.; Hanakawa, Y.; Nishifuji, K.; Sugai, M.; Stanley, J.R. Staphylococcal exfoliative toxin B specifically cleaves desmoglein 1. J. Investig. Dermatol. 2002, 118, 845–850.
- Oliveira, D.; Borges, A.; Simões, M. Staphylococcus aureus Toxins and Their Molecular Activity in Infectious Diseases. Toxins 2018, 10, 252.
- Nordengrün, M.; Abdurrahman, G.; Treffon, J.; Wächter, H.; Kahl, B.C.; Bröker, B.M. Allergic Reactions to Serine Protease-Like Proteins of Staphylococcus aureus. Front. Immunol. 2021, 12, 651060.
- Smagur, J.; Guzik, K.; Magiera, L.; Bzowska, M.; Gruca, M.; Thøgersen, I.B.; Enghild, J.J.; Potempa, J. A new pathway of staphylococcal pathogenesis: Apoptosis-like death induced by Staphopain B in human neutrophils and monocytes. J. Innate Immun. 2009, 1, 98–108.
- Kulig, P.; Zabel, B.A.; Dubin, G.; Allen, S.J.; Ohyama, T.; Potempa, J.; Handel, T.M.; Butcher, E.C.; Cichy, J. Staphylococcus aureus-derived staphopain B, a potent cysteine protease activator of plasma chemerin. J. Immunol. 2007, 178, 3713–3720.
- Bergdoll, M.S.; Crass, B.A.; Reiser, R.F.; Robbins, R.N.; Davis, J.P. A new staphylococcal enterotoxin, enterotoxin F, associated with toxic-shock-syndrome Staphylococcus aureus isolates. Lancet 1981, 1, 1017–1021.
- McCormick, J.K.; Yarwood, J.M.; Schlievert, P.M. Toxic shock syndrome and bacterial superantigens: An update. Annu Rev. Microbiol. 2001, 55, 77–104.
- Calus, L.; Derycke, L.; Dullaers, M.; Van Zele, T.; De Ruyck, N.; Pérez-Novo, C.; Holtappels, G.; De Vos, G.; Lambrecht, B.N.; Bachert, C.; et al. IL-21 Is Increased in Nasal Polyposis and after Stimulation with Staphylococcus aureus Enterotoxin B. Int. Arch. Allergy Immunol. 2017, 174, 161–169.
- Bauquet, A.T.; Jin, H.; Paterson, A.M.; Mitsdoerffer, M.; Ho, I.C.; Sharpe, A.H.; Kuchroo, V.K. The costimulatory molecule ICOS regulates the expression of c-Maf and IL-21 in the development of follicular T helper cells and TH-17 cells. Nat. Immunol. 2009, 10, 167–175.
- Van Zele, T.; Gevaert, P.; Watelet, J.B.; Claeys, G.; Holtappels, G.; Claeys, C.; van Cauwenberge, P.; Bachert, C. Staphylococcus aureus colonization and IgE antibody formation to enterotoxins is increased in nasal polyposis. J. Allergy Clin. Immunol. 2004, 114, 981–983.
- Orfali, R.L.; da Silva Oliveira, L.M.; de Lima, J.F.; de Carvalho, G.C.; Ramos, Y.A.L.; Pereira, N.Z.; Pereira, N.V.; Zaniboni, M.C.; Sotto, M.N.; da Silva Duarte, A.J.; et al. Staphylococcus aureus enterotoxins modulate IL-22-secreting cells in adults with atopic dermatitis. Sci. Rep. 2018, 8, 6665.
- Hellings, P.W.; Hens, G.; Meyts, I.; Bullens, D.; Vanoirbeek, J.; Gevaert, P.; Jorissen, M.; Ceuppens, J.L.; Bachert, C. Aggravation of bronchial eosinophilia in mice by nasal and bronchial exposure to Staphylococcus aureus enterotoxin B. Clin. Exp. Allergy 2006, 36, 1063–1071.
- Liu, X.; Wen, Y.; Wang, D.; Zhao, Z.; Jeffry, J.; Zeng, L.; Zou, Z.; Chen, H.; Tao, A. Synergistic activation of Src, ERK and STAT pathways in PBMCs for Staphylococcal enterotoxin A induced production of cytokines and chemokines. Asian Pac. J. Allergy Immunol. 2020, 38, 52–63.
- Naik, S.; Smith, F.; Ho, J.; Croft, N.M.; Domizio, P.; Price, E.; Sanderson, I.R.; Meadows, N.J. Staphylococcal enterotoxins G and I, a cause of severe but reversible neonatal enteropathy. Clin. Gastroenterol. Hepatol. 2008, 6, 251–254.
- Liu, Y.; Chen, W.; Ali, T.; Alkasir, R.; Yin, J.; Liu, G.; Han, B. Staphylococcal enterotoxin H induced apoptosis of bovine mammary epithelial cells in vitro. Toxins 2014, 6, 3552–3567.
- Hou, F.; Peng, L.; Jiang, J.; Chen, T.; Xu, D.; Huang, Q.; Ye, C.; Peng, Y.; Hu, D.L.; Fang, R. ATP Facilitates Staphylococcal Enterotoxin O Induced Neutrophil IL-1β Secretion via NLRP3 Inflammasome Dependent Pathways. Front. Immunol. 2021, 12, 649235.
- Zhao, Y.; Tang, J.; Yang, D.; Tang, C.; Chen, J. Staphylococcal enterotoxin M induced inflammation and impairment of bovine mammary epithelial cells. J. Dairy Sci. 2020, 103, 8350–8359.
- Zhao, Y.; Zhu, A.; Tang, J.; Tang, C.; Chen, J. Identification and measurement of staphylococcal enterotoxin M from Staphylococcus aureus isolate associated with staphylococcal food poisoning. Lett. Appl. Microbiol. 2017, 65, 27–34.
- Hu, D.L.; Ono, H.K.; Isayama, S.; Okada, R.; Okamura, M.; Lei, L.C.; Liu, Z.S.; Zhang, X.C.; Liu, M.Y.; Cui, J.C.; et al. Biological characteristics of staphylococcal enterotoxin Q and its potential risk for food poisoning. J. Appl. Microbiol. 2017, 122, 1672–1679.
- Tian, X.; Huang, Q.; Liang, J.; Wang, J.; Zhang, J.; Yang, Y.; Ye, Q.; He, S.; Li, J.; Wu, Z.; et al. A review of the mechanisms of keratinocytes damage caused by Staphylococcus aureus infection in patients with atopic dermatitis. J. Leukoc. Biol. 2021, 110, 1163–1169.
- Cruciani, M.; Etna, M.P.; Camilli, R.; Giacomini, E.; Percario, Z.A.; Severa, M.; Sandini, S.; Rizzo, F.; Brandi, V.; Balsamo, G.; et al. Staphylococcus aureus Esx Factors Control Human Dendritic Cell Functions Conditioning Th1/Th17 Response. Front. Cell. Infect. Microbiol. 2017, 7, 330.
- Korea, C.G.; Balsamo, G.; Pezzicoli, A.; Merakou, C.; Tavarini, S.; Bagnoli, F.; Serruto, D.; Unnikrishnan, M. Staphylococcal Esx proteins modulate apoptosis and release of intracellular Staphylococcus aureus during infection in epithelial cells. Infect. Immun. 2014, 82, 4144–4153.
- Becker, K.; Heilmann, C.; Peters, G. Coagulase-negative staphylococci. Clin. Microbiol. Rev. 2014, 27, 870–926.
- Jin, T.; Zhu, Y.L.; Li, J.; Shi, J.; He, X.Q.; Ding, J.; Xu, Y.Q. Staphylococcal protein A, Panton-Valentine leukocidin and coagulase aggravate the bone loss and bone destruction in osteomyelitis. Cell Physiol. Biochem. 2013, 32, 322–333.
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535.
- Berends, E.T.; Horswill, A.R.; Haste, N.M.; Monestier, M.; Nizet, V.; von Köckritz-Blickwede, M. Nuclease expression by Staphylococcus aureus facilitates escape from neutrophil extracellular traps. J. Innate Immun. 2010, 2, 576–586.
- Thammavongsa, V.; Missiakas, D.M.; Schneewind, O. Staphylococcus aureus degrades neutrophil extracellular traps to promote immune cell death. Science 2013, 342, 863–866.
- Winstel, V.; Schneewind, O.; Missiakas, D. Staphylococcus aureus Exploits the Host Apoptotic Pathway To Persist during Infection. mBio 2019, 10, e02270-19.
- Wang, H.; von Rohrscheidt, J.; Roehrbein, J.; Peters, T.; Sindrilaru, A.; Kess, D.; Preissner, K.T.; Scharffetter-Kochanek, K. Extracellular adherence protein of Staphylococcus aureus suppresses disease by inhibiting T-cell recruitment in a mouse model of psoriasis. J. Investig. Dermatol. 2010, 130, 743–754.
- Foster, T.J. The MSCRAMM Family of Cell-Wall-Anchored Surface Proteins of Gram-Positive Cocci. Trends Microbiol. 2019, 27, 927–941.
- Pazos, M.; Peters, K. Peptidoglycan. Subcell. Biochem. 2019, 92, 127–168.
- Foster, T.J. Surface Proteins of Staphylococcus aureus. Microbiol. Spectr. 2019, 7, 4.
- Rivas, J.M.; Speziale, P.; Patti, J.M.; Höök, M. MSCRAMM--targeted vaccines and immunotherapy for staphylococcal infection. Curr. Opin. Drug Discov. Dev. 2004, 7, 223–227.
- Fox, P.G.; Schiavetti, F.; Rappuoli, R.; McLoughlin, R.M.; Bagnoli, F. Staphylococcal Protein A Induces Leukocyte Necrosis by Complexing with Human Immunoglobulins. mBio 2021, 12, e00899-21.
- Das, T.; Sa, G.; Chattopadhyay, S.; Ray, P.K. Protein A-induced apoptosis of cancer cells is effected by soluble immune mediators. Cancer Immunol. Immunother. 2002, 51, 376–380.
- Sawada, M.; Nakashima, S.; Banno, Y.; Yamakawa, H.; Takenaka, K.; Shinoda, J.; Nishimura, Y.; Sakai, N.; Nozawa, Y. Influence of Bax or Bcl-2 overexpression on the ceramide-dependent apoptotic pathway in glioma cells. Oncogene 2000, 19, 3508–3520.
- Claro, T.; Widaa, A.; McDonnell, C.; Foster, T.J.; O’Brien, F.J.; Kerrigan, S.W. Staphylococcus aureus protein A binding to osteoblast tumour necrosis factor receptor 1 results in activation of nuclear factor kappa B and release of interleukin-6 in bone infection. Microbiology 2013, 159, 147–154.
- Claro, T.; Widaa, A.; O’Seaghdha, M.; Miajlovic, H.; Foster, T.J.; O’Brien, F.J.; Kerrigan, S.W. Staphylococcus aureus protein A binds to osteoblasts and triggers signals that weaken bone in osteomyelitis. PLoS ONE 2011, 6, e18748.
- Al Kindi, A.; Williams, H.; Matsuda, K.; Alkahtani, A.M.; Saville, C.; Bennett, H.; Alshammari, Y.; Tan, S.Y.; O’Neill, C.; Tanaka, A.; et al. Staphylococcus aureus second immunoglobulin-binding protein drives atopic dermatitis via IL-33. J. Allergy Clin. Immunol. 2021, 147, 1354–1368.e3.
- Garcovich, S.; Maurelli, M.; Gisondi, P.; Peris, K.; Yosipovitch, G.; Girolomoni, G. Pruritus as a Distinctive Feature of Type 2 Inflammation. Vaccines 2021, 9, 303.
- Imai, Y. Interleukin-33 in atopic dermatitis. J. Dermatol. Sci. 2019, 96, 2–7.
- Takeda, K.; Takeuchi, O.; Akira, S. Recognition of lipopeptides by Toll-like receptors. J. Endotoxin. Res. 2002, 8, 459–463.
- Stoll, H.; Dengjel, J.; Nerz, C.; Götz, F. Staphylococcus aureus deficient in lipidation of prelipoproteins is attenuated in growth and immune activation. Infect. Immun. 2005, 73, 2411–2423.
- Takeuchi, O.; Kawai, T.; Mühlradt, P.F.; Morr, M.; Radolf, J.D.; Zychlinsky, A.; Takeda, K.; Akira, S. Discrimination of bacterial lipoproteins by Toll-like receptor 6. Int. Immunol. 2001, 13, 933–940.
- Hara, H.; Seregin, S.S.; Yang, D.; Fukase, K.; Chamaillard, M.; Alnemri, E.S.; Inohara, N.; Chen, G.Y.; Núñez, G. The NLRP6 Inflammasome Recognizes Lipoteichoic Acid and Regulates Gram-Positive Pathogen Infection. Cell 2018, 175, 1651–1664.e14.
- Hattar, K.; Grandel, U.; Moeller, A.; Fink, L.; Iglhaut, J.; Hartung, T.; Morath, S.; Seeger, W.; Grimminger, F.; Sibelius, U. Lipoteichoic acid (LTA) from Staphylococcus aureus stimulates human neutrophil cytokine release by a CD14-dependent, Toll-like-receptor-independent mechanism: Autocrine role of tumor necrosis factor- in mediating LTA-induced interleukin-8 generation. Crit. Care Med. 2006, 34, 835–841.
- Brauweiler, A.M.; Goleva, E.; Leung, D.Y.M. Staphylococcus aureus Lipoteichoic Acid Initiates a TSLP-Basophil-IL4 Axis in the Skin. J. Investig. Dermatol. 2020, 140, 915–917.e2.
- Misawa, Y.; Kelley, K.A.; Wang, X.; Wang, L.; Park, W.B.; Birtel, J.; Saslowsky, D.; Lee, J.C. Staphylococcus aureus Colonization of the Mouse Gastrointestinal Tract Is Modulated by Wall Teichoic Acid, Capsule, and Surface Proteins. PLoS Pathog. 2015, 11, e1005061.
- Pasquina-Lemonche, L.; Burns, J.; Turner, R.D.; Kumar, S.; Tank, R.; Mullin, N.; Wilson, J.S.; Chakrabarti, B.; Bullough, P.A.; Foster, S.J.; et al. The architecture of the Gram-positive bacterial cell wall. Nature 2020, 582, 294–297.
- Covas, G.; Vaz, F.; Henriques, G.; Pinho, M.G.; Filipe, S.R. Analysis of Cell Wall Teichoic Acids in Staphylococcus aureus. Methods Mol. Biol. 2016, 1440, 201–213.
- Arroyo, D.S.; Soria, J.A.; Gaviglio, E.A.; Garcia-Keller, C.; Cancela, L.M.; Rodriguez-Galan, M.C.; Wang, J.M.; Iribarren, P. Toll-like receptor 2 ligands promote microglial cell death by inducing autophagy. FASEB J. 2013, 27, 299–312.
- Matsui, K.; Tofukuji, S.; Ikeda, R. CCL17 production by mouse langerhans cells stimulated with Staphylococcus aureus cell wall components. Biol. Pharm. Bull. 2015, 38, 317–320.
- Matsui, K.; Wirotesangthong, M.; Nishikawa, A. Peptidoglycan from Staphylococcus aureus induces IL-4 production from murine spleen cells via an IL-18-dependent mechanism. Int. Arch. Allergy Immunol. 2008, 146, 262–266.
- Supajatura, V.; Ushio, H.; Nakao, A.; Akira, S.; Okumura, K.; Ra, C.; Ogawa, H. Differential responses of mast cell Toll-like receptors 2 and 4 in allergy and innate immunity. J. Clin. Investig. 2002, 109, 1351–1359.
- Biggs, T.C.; Abadalkareem, R.S.; Hayes, S.M.; Holding, R.E.; Lau, L.C.; Harries, P.G.; Allan, R.N.; Pender, S.L.F.; Walls, A.F.; Salib, R.J. Staphylococcus aureus internalisation enhances bacterial survival through modulation of host immune responses and mast cell activation. Allergy 2021, 76, 1893–1896.
- Lin, H.Y.; Tang, C.H.; Chen, J.H.; Chuang, J.Y.; Huang, S.M.; Tan, T.W.; Lai, C.H.; Lu, D.Y. Peptidoglycan induces interleukin-6 expression through the TLR2 receptor, JNK, c-Jun, and AP-1 pathways in microglia. J. Cell. Physiol. 2011, 226, 1573–1582.
- Hsu, M.J.; Chang, C.K.; Chen, M.C.; Chen, B.C.; Ma, H.P.; Hong, C.Y.; Lin, C.H. Apoptosis signal-regulating kinase 1 in peptidoglycan-induced COX-2 expression in macrophages. J. Leukoc. Biol. 2010, 87, 1069–1082.
- Wang, D.; Xiao, P.L.; Duan, H.X.; Zhou, M.; Liu, J.; Li, W.; Luo, K.L.; Chen, J.J.; Hu, J.Y. Peptidoglycans promotes human leukemic THP-1 cell apoptosis and differentiation. Asian Pac. J. Cancer Prev. 2012, 13, 6409–6413.
- Namazi, M.R. Paradoxical exacerbation of psoriasis in AIDS: Proposed explanations including the potential roles of substance P and gram-negative bacteria. Autoimmunity 2004, 37, 67–71.
- Ruíz-González, V.; Cancino-Diaz, J.C.; Rodríguez-Martínez, S.; Cancino-Diaz, M.E. Keratinocytes treated with peptidoglycan from Staphylococcus aureus produce vascular endothelial growth factor, and its expression is amplified by the subsequent production of interleukin-13. Int. J. Dermatol. 2009, 48, 846–854.
- Wu, H.M.; Wang, J.; Zhang, B.; Fang, L.; Xu, K.; Liu, R.Y. CpG-ODN promotes phagocytosis and autophagy through JNK/P38 signal pathway in Staphylococcus aureus-stimulated macrophage. Life Sci. 2016, 161, 51–59.
- Müller, S.; Wolf, A.J.; Iliev, I.D.; Berg, B.L.; Underhill, D.M.; Liu, G.Y. Poorly Cross-Linked Peptidoglycan in MRSA Due to mecA Induction Activates the Inflammasome and Exacerbates Immunopathology. Cell Host Microbe 2015, 18, 604–612.
- Zhu, F.; Zhou, Y.; Jiang, C.; Zhang, X. Role of JAK-STAT signaling in maturation of phagosomes containing Staphylococcus aureus. Sci. Rep. 2015, 5, 14854.
- Shi, M.; Willing, S.E.; Kim, H.K.; Schneewind, O.; Missiakas, D. Peptidoglycan Contribution to the B Cell Superantigen Activity of Staphylococcal Protein A. mBio 2021, 12, e00039-21.
- Moriwaki, M.; Iwamoto, K.; Niitsu, Y.; Matsushima, A.; Yanase, Y.; Hisatsune, J.; Sugai, M.; Hide, M. Staphylococcus aureus from atopic dermatitis skin accumulates in the lysosomes of keratinocytes with induction of IL-1α secretion via TLR9. Allergy 2019, 74, 560–571.
- Wang, G.; Sweren, E.; Liu, H.; Wier, E.; Alphonse, M.P.; Chen, R.; Islam, N.; Li, A.; Xue, Y.; Chen, J.; et al. Bacteria induce skin regeneration via IL-1β signaling. Cell Host Microbe 2021, 29, 777–791.e6.
- Cayrol, C.; Girard, J.P. Interleukin-33 (IL-33): A nuclear cytokine from the IL-1 family. Immunol. Rev. 2018, 281, 154–168.
- Liu, B.; Tai, Y.; Achanta, S.; Kaelberer, M.M.; Caceres, A.I.; Shao, X.; Fang, J.; Jordt, S.E. IL-33/ST2 signaling excites sensory neurons and mediates itch response in a mouse model of poison ivy contact allergy. Proc. Natl. Acad. Sci. USA 2016, 113, E7572–E7579.
- Vu, A.T.; Baba, T.; Chen, X.; Le, T.A.; Kinoshita, H.; Xie, Y.; Kamijo, S.; Hiramatsu, K.; Ikeda, S.; Ogawa, H.; et al. Staphylococcus aureus membrane and diacylated lipopeptide induce thymic stromal lymphopoietin in keratinocytes through the Toll-like receptor 2-Toll-like receptor 6 pathway. J. Allergy Clin. Immunol. 2010, 126, 985–993.e3.
- Wilson, S.R.; Thé, L.; Batia, L.M.; Beattie, K.; Katibah, G.E.; McClain, S.P.; Pellegrino, M.; Estandian, D.M.; Bautista, D.M. The epithelial cell-derived atopic dermatitis cytokine TSLP activates neurons to induce itch. Cell 2013, 155, 285–295.
- Son, E.D.; Kim, H.J.; Park, T.; Shin, K.; Bae, I.H.; Lim, K.M.; Cho, E.G.; Lee, T.R. Staphylococcus aureus inhibits terminal differentiation of normal human keratinocytes by stimulating interleukin-6 secretion. J. Dermatol. Sci. 2014, 74, 64–71.
- Williams, M.R.; Nakatsuji, T.; Sanford, J.A.; Vrbanac, A.F.; Gallo, R.L. Staphylococcus aureus Induces Increased Serine Protease Activity in Keratinocytes. J. Investig. Dermatol. 2017, 137, 377–384.
- Lan, F.; Zhang, N.; Holtappels, G.; De Ruyck, N.; Krysko, O.; Van Crombruggen, K.; Braun, H.; Johnston, S.L.; Papadopoulos, N.G.; Zhang, L.; et al. Staphylococcus aureus Induces a Mucosal Type 2 Immune Response via Epithelial Cell-derived Cytokines. Am. J. Respir. Crit Care Med. 2018, 198, 452–463.
- Hardman, C.S.; Chen, Y.L.; Salimi, M.; Nahler, J.; Corridoni, D.; Jagielowicz, M.; Fonseka, C.L.; Johnson, D.; Repapi, E.; Cousins, D.J.; et al. IL-6 effector function of group 2 innate lymphoid cells (ILC2) is NOD2 dependent. Sci. Immunol. 2021, 6, eabe5084.
- Romagnani, S. T-cell subsets (Th1 versus Th2). Ann. Allergy Asthma Immunol. 2000, 85, 9–18; quiz 18, 21.
- Pérez Novo, C.A.; Jedrzejczak-Czechowicz, M.; Lewandowska-Polak, A.; Claeys, C.; Holtappels, G.; Van Cauwenberge, P.; Kowalski, M.L.; Bachert, C. T cell inflammatory response, Foxp3 and TNFRS18-L regulation of peripheral blood mononuclear cells from patients with nasal polyps-asthma after staphylococcal superantigen stimulation. Clin. Exp. Allergy 2010, 40, 1323–1332.
- Orciani, M.; Campanati, A.; Caffarini, M.; Ganzetti, G.; Consales, V.; Lucarini, G.; Offidani, A.; Di Primio, R. T helper (Th)1, Th17 and Th2 imbalance in mesenchymal stem cells of adult patients with atopic dermatitis: At the origin of the problem. Br. J. Dermatol. 2017, 176, 1569–1576.
- Tada, Y.; Asahina, A.; Takekoshi, T.; Kishimoto, E.; Mitsui, H.; Saeki, H.; Komine, M.; Tamaki, K. Interleukin 12 production by monocytes from patients with psoriasis and its inhibition by ciclosporin A. Br. J. Dermatol. 2006, 154, 1180–1183.
- Stetson, D.B.; Voehringer, D.; Grogan, J.L.; Xu, M.; Reinhardt, R.L.; Scheu, S.; Kelly, B.L.; Locksley, R.M. Th2 cells: Orchestrating barrier immunity. Adv. Immunol. 2004, 83, 163–189.
- Kamijo, H.; Miyagaki, T.; Hayashi, Y.; Akatsuka, T.; Watanabe-Otobe, S.; Oka, T.; Shishido-Takahashi, N.; Suga, H.; Sugaya, M.; Sato, S. Increased IL-26 Expression Promotes T Helper Type 17- and T Helper Type 2-Associated Cytokine Production by Keratinocytes in Atopic Dermatitis. J. Investig. Dermatol. 2020, 140, 636–644.e2.
- Chang, H.W.; Yan, D.; Singh, R.; Liu, J.; Lu, X.; Ucmak, D.; Lee, K.; Afifi, L.; Fadrosh, D.; Leech, J.; et al. Alteration of the cutaneous microbiome in psoriasis and potential role in Th17 polarization. Microbiome 2018, 6, 154.
- Brauweiler, A.M.; Goleva, E.; Leung, D.Y.M. Th2 cytokines increase Staphylococcus aureus alpha toxin-induced keratinocyte death through the signal transducer and activator of transcription 6 (STAT6). J. Investig. Dermatol. 2014, 134, 2114–2121.
- Ou, L.S.; Goleva, E.; Hall, C.; Leung, D.Y. T regulatory cells in atopic dermatitis and subversion of their activity by superantigens. J. Allergy Clin. Immunol. 2004, 113, 756–763.
- Laouini, D.; Kawamoto, S.; Yalcindag, A.; Bryce, P.; Mizoguchi, E.; Oettgen, H.; Geha, R.S. Epicutaneous sensitization with superantigen induces allergic skin inflammation. J. Allergy Clin. Immunol. 2003, 112, 981–987.
- Jacobsen, E.A.; Ochkur, S.I.; Lee, N.A.; Lee, J.J. Eosinophils and asthma. Curr. Allergy Asthma Rep. 2007, 7, 18–26.
- Fujieda, S.; Imoto, Y.; Kato, Y.; Ninomiya, T.; Tokunaga, T.; Tsutsumiuchi, T.; Yoshida, K.; Kidoguchi, M.; Takabayashi, T. Eosinophilic chronic rhinosinusitis. Allergol. Int. 2019, 68, 403–412.
- Warner, J.A.; McGirt, L.Y.; Beck, L.A. Biomarkers of Th2 polarity are predictive of staphylococcal colonization in subjects with atopic dermatitis. Br. J. Dermatol. 2009, 160, 183–185.
- Dubin, C.; Del Duca, E.; Guttman-Yassky, E. The IL-4, IL-13 and IL-31 pathways in atopic dermatitis. Expert Rev. Clin. Immunol. 2021, 17, 835–852.
- Iwaszko, M.; Biały, S.; Bogunia-Kubik, K. Significance of Interleukin (IL)-4 and IL-13 in Inflammatory Arthritis. Cells 2021, 10, 3000.
- Kimura, A.; Kishimoto, T. Th17 cells in inflammation. Int. Immunopharmacol 2011, 11, 319–322.
- Tokura, Y. Th17 cells and skin diseases. Nihon Rinsho Meneki Gakkai Kaishi 2012, 35, 388–392.
- Koga, C.; Kabashima, K.; Shiraishi, N.; Kobayashi, M.; Tokura, Y. Possible pathogenic role of Th17 cells for atopic dermatitis. J. Investig. Dermatol. 2008, 128, 2625–2630.
- Orfali, R.L.; Yoshikawa, F.S.Y.; Oliveira, L.; Pereira, N.Z.; de Lima, J.F.; Ramos YÁ, L.; Duarte, A.; Sato, M.N.; Aoki, V. Staphylococcal enterotoxins modulate the effector CD4(+) T cell response by reshaping the gene expression profile in adults with atopic dermatitis. Sci. Rep. 2019, 9, 13082.
- Sugaya, M. The Role of Th17-Related Cytokines in Atopic Dermatitis. Int. J. Mol. Sci. 2020, 21, 1314.
- Liu, H.; Archer, N.K.; Dillen, C.A.; Wang, Y.; Ashbaugh, A.G.; Ortines, R.V.; Kao, T.; Lee, S.K.; Cai, S.S.; Miller, R.J.; et al. Staphylococcus aureus Epicutaneous Exposure Drives Skin Inflammation via IL-36-Mediated T Cell Responses. Cell Host Microbe 2017, 22, 653–666.e5.
- Dey, I.; Bishayi, B. Role of Th17 and Treg cells in septic arthritis and the impact of the Th17/Treg-derived cytokines in the pathogenesis of S. aureus induced septic arthritis in mice. Microb. Pathog. 2017, 113, 248–264.
- Dey, I.; Bishayi, B. Role of different Th17 and Treg downstream signalling pathways in the pathogenesis of Staphylococcus aureus infection induced septic arthritis in mice. Exp. Mol. Pathol. 2020, 116, 104485.
- Lee, G.R. The Balance of Th17 versus Treg Cells in Autoimmunity. Int. J. Mol. Sci. 2018, 19, 730.
- Sultana, S.; Dey, R.; Bishayi, B. Dual neutralization of TNFR-2 and MMP-2 regulates the severity of induced septic arthritis correlating alteration in the level of interferon gamma and interleukin-10 in terms of TNFR2 blocking. Immunol. Res. 2018, 66, 97–119.
- Ghosh, R.; Dey, R.; Sawoo, R.; Bishayi, B. Neutralization of IL-17 and treatment with IL-2 protects septic arthritis by regulating free radical production and antioxidant enzymes in Th17 and Tregs: An immunomodulatory TLR2 versus TNFR response. Cell Immunol. 2021, 370, 104441.
- Saito, S.; Quadery, A.F. Staphylococcus aureus Lipoprotein Induces Skin Inflammation, Accompanied with IFN-γ-Producing T Cell Accumulation through Dermal Dendritic Cells. Pathogens 2018, 7, 64.
- Taylor, A.L.; Llewelyn, M.J. Superantigen-induced proliferation of human CD4+CD25-T cells is followed by a switch to a functional regulatory phenotype. J. Immunol. 2010, 185, 6591–6598.
- Seneschal, J.; Clark, R.A.; Gehad, A.; Baecher-Allan, C.M.; Kupper, T.S. Human epidermal Langerhans cells maintain immune homeostasis in skin by activating skin resident regulatory T cells. Immunity 2012, 36, 873–884.
- van Dalen, R.; De La Cruz Diaz, J.S.; Rumpret, M.; Fuchsberger, F.F.; van Teijlingen, N.H.; Hanske, J.; Rademacher, C.; Geijtenbeek, T.B.H.; van Strijp, J.A.G.; Weidenmaier, C.; et al. Langerhans Cells Sense Staphylococcus aureus Wall Teichoic Acid through Langerin To Induce Inflammatory Responses. mBio 2019, 10, e00330-19.
- Ma, L.; Xue, H.B.; Guan, X.H.; Shu, C.M.; Wang, F.; Zhang, J.H.; An, R.Z. The Imbalance of Th17 cells and CD4(+) CD25(high) Foxp3(+) Treg cells in patients with atopic dermatitis. J. Eur. Acad. Dermatol. Venereol. 2014, 28, 1079–1086.
- Crotty, S. T follicular helper cell differentiation, function, and roles in disease. Immunity 2014, 41, 529–542.
- Moser, B. CXCR5, the Defining Marker for Follicular B Helper T (TFH) Cells. Front. Immunol. 2015, 6, 296.
- Bossaller, L.; Burger, J.; Draeger, R.; Grimbacher, B.; Knoth, R.; Plebani, A.; Durandy, A.; Baumann, U.; Schlesier, M.; Welcher, A.A.; et al. ICOS deficiency is associated with a severe reduction of CXCR5+CD4 germinal center Th cells. J. Immunol. 2006, 177, 4927–4932.
- Bennett, F.; Luxenberg, D.; Ling, V.; Wang, I.M.; Marquette, K.; Lowe, D.; Khan, N.; Veldman, G.; Jacobs, K.A.; Valge-Archer, V.E.; et al. Program death-1 engagement upon TCR activation has distinct effects on costimulation and cytokine-driven proliferation: Attenuation of ICOS, IL-4, and IL-21, but not CD28, IL-7, and IL-15 responses. J. Immunol. 2003, 170, 711–718.
- Sage, P.T.; Paterson, A.M.; Lovitch, S.B.; Sharpe, A.H. The coinhibitory receptor CTLA-4 controls B cell responses by modulating T follicular helper, T follicular regulatory, and T regulatory cells. Immunity 2014, 41, 1026–1039.
- Johnston, R.J.; Poholek, A.C.; DiToro, D.; Yusuf, I.; Eto, D.; Barnett, B.; Dent, A.L.; Craft, J.; Crotty, S. Bcl6 and Blimp-1 are reciprocal and antagonistic regulators of T follicular helper cell differentiation. Science 2009, 325, 1006–1010.
- Dupont, C.D.; Scully, I.L.; Zimnisky, R.M.; Monian, B.; Rossitto, C.P.; O’Connell, E.B.; Jansen, K.U.; Anderson, A.S.; Love, J.C. Two Vaccines for Staphylococcus aureus Induce a B-Cell-Mediated Immune Response. mSphere 2018, 3, e00217-18.
- Chang, H.C.; Sehra, S.; Goswami, R.; Yao, W.; Yu, Q.; Stritesky, G.L.; Jabeen, R.; McKinley, C.; Ahyi, A.N.; Han, L.; et al. The transcription factor PU.1 is required for the development of IL-9-producing T cells and allergic inflammation. Nat. Immunol. 2010, 11, 527–534.
- Soussi-Gounni, A.; Kontolemos, M.; Hamid, Q. Role of IL-9 in the pathophysiology of allergic diseases. J. Allergy Clin. Immunol. 2001, 107, 575–582.
- Jäger, A.; Dardalhon, V.; Sobel, R.A.; Bettelli, E.; Kuchroo, V.K. Th1, Th17, and Th9 effector cells induce experimental autoimmune encephalomyelitis with different pathological phenotypes. J. Immunol. 2009, 183, 7169–7177.
- Purwar, R.; Schlapbach, C.; Xiao, S.; Kang, H.S.; Elyaman, W.; Jiang, X.; Jetten, A.M.; Khoury, S.J.; Fuhlbrigge, R.C.; Kuchroo, V.K.; et al. Robust tumor immunity to melanoma mediated by interleukin-9-producing T cells. Nat. Med. 2012, 18, 1248–1253.
- Vivier, E.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.J.; Mebius, R.E.; et al. Innate Lymphoid Cells: 10 Years On. Cell 2018, 174, 1054–1066.
- Ghaedi, M.; Takei, F. Innate lymphoid cell development. J. Allergy Clin. Immunol. 2021, 147, 1549–1560.
- Annunziato, F.; Romagnani, C.; Romagnani, S. The 3 major types of innate and adaptive cell-mediated effector immunity. J. Allergy Clin. Immunol. 2015, 135, 626–635.
- Gentek, R.; Molawi, K.; Sieweke, M.H. Tissue macrophage identity and self-renewal. Immunol. Rev. 2014, 262, 56–73.
- Pidwill, G.R.; Gibson, J.F.; Cole, J.; Renshaw, S.A.; Foster, S.J. The Role of Macrophages in Staphylococcus aureus Infection. Front. Immunol. 2020, 11, 620339.
- Thurlow, L.R.; Hanke, M.L.; Fritz, T.; Angle, A.; Aldrich, A.; Williams, S.H.; Engebretsen, I.L.; Bayles, K.W.; Horswill, A.R.; Kielian, T. Staphylococcus aureus biofilms prevent macrophage phagocytosis and attenuate inflammation in vivo. J. Immunol. 2011, 186, 6585–6596.
- Wang, X.; Eagen, W.J.; Lee, J.C. Orchestration of human macrophage NLRP3 inflammasome activation by Staphylococcus aureus extracellular vesicles. Proc. Natl. Acad. Sci. USA 2020, 117, 3174–3184.
- Flannagan, R.S.; Heinrichs, D.E. Macrophage-driven nutrient delivery to phagosomal Staphylococcus aureus supports bacterial growth. EMBO Rep. 2020, 21, e50348.
- Grayczyk, J.P.; Alonzo, F., 3rd. Staphylococcus aureus Lipoic Acid Synthesis Limits Macrophage Reactive Oxygen and Nitrogen Species Production To Promote Survival during Infection. Infect. Immun. 2019, 87, e00344-19.
- Tatsuno, K.; Fujiyama, T.; Yamaguchi, H.; Waki, M.; Tokura, Y. TSLP Directly Interacts with Skin-Homing Th2 Cells Highly Expressing its Receptor to Enhance IL-4 Production in Atopic Dermatitis. J. Investig. Dermatol. 2015, 135, 3017–3024.
- Iwamoto, K.; Nümm, T.J.; Koch, S.; Herrmann, N.; Leib, N.; Bieber, T. Langerhans and inflammatory dendritic epidermal cells in atopic dermatitis are tolerized toward TLR2 activation. Allergy 2018, 73, 2205–2213.
- Asahina, A.; Tamaki, K. Role of Langerhans cells in cutaneous protective immunity: Is the reappraisal necessary? J. Dermatol. Sci. 2006, 44, 1–9.
- Berends, E.T.M.; Zheng, X.; Zwack, E.E.; Ménager, M.M.; Cammer, M.; Shopsin, B.; Torres, V.J. Staphylococcus aureus Impairs the Function of and Kills Human Dendritic Cells via the LukAB Toxin. mBio 2019, 10, e01918-18.
- Matsui, K.; Kanai, S.; Ikuta, M.; Horikawa, S. Mast Cells Stimulated with Peptidoglycan from Staphylococcus aureus Augment the Development of Th1 Cells. J. Pharm. Pharm. Sci. 2018, 21, 296–304.
- Hayes, S.M.; Biggs, T.C.; Goldie, S.P.; Harries, P.G.; Walls, A.F.; Allan, R.N.; Pender, S.L.F.; Salib, R.J. Staphylococcus aureus internalization in mast cells in nasal polyps: Characterization of interactions and potential mechanisms. J. Allergy Clin. Immunol. 2020, 145, 147–159.
- McFadden, J.P.; Noble, W.C.; Camp, R.D. Superantigenic exotoxin-secreting potential of staphylococci isolated from atopic eczematous skin. Br. J. Dermatol. 1993, 128, 631–632.
- Geoghegan, J.A.; Irvine, A.D.; Foster, T.J. Staphylococcus aureus and Atopic Dermatitis: A Complex and Evolving Relationship. Trends Microbiol. 2018, 26, 484–497.
- Bachert, C.; Humbert, M.; Hanania, N.A.; Zhang, N.; Holgate, S.; Buhl, R.; Bröker, B.M. Staphylococcus aureus and its IgE-inducing enterotoxins in asthma: Current knowledge. Eur. Respir. J. 2020, 55, 1901592.
- Liu, C.; Yang, L.; Han, Y.; Ouyang, W.; Yin, W.; Xu, F. Mast cells participate in regulation of lung-gut axis during Staphylococcus aureus pneumonia. Cell Prolif. 2019, 52, e12565.
- Lehman, H.K.; Segal, B.H. The role of neutrophils in host defense and disease. J. Allergy Clin. Immunol. 2020, 145, 1535–1544.
- Gough, P.; Ganesan, S.; Datta, S.K. IL-20 Signaling in Activated Human Neutrophils Inhibits Neutrophil Migration and Function. J. Immunol. 2017, 198, 4373–4382.
- Marchitto, M.C.; Dillen, C.A.; Liu, H.; Miller, R.J.; Archer, N.K.; Ortines, R.V.; Alphonse, M.P.; Marusina, A.I.; Merleev, A.A.; Wang, Y.; et al. Clonal Vγ6(+)Vδ4(+) T cells promote IL-17-mediated immunity against Staphylococcus aureus skin infection. Proc. Natl. Acad. Sci. USA 2019, 116, 10917–10926.
- Cho, J.S.; Guo, Y.; Ramos, R.I.; Hebroni, F.; Plaisier, S.B.; Xuan, C.; Granick, J.L.; Matsushima, H.; Takashima, A.; Iwakura, Y.; et al. Neutrophil-derived IL-1β is sufficient for abscess formation in immunity against Staphylococcus aureus in mice. PLoS Pathog. 2012, 8, e1003047.
- Lutalo, P.M.; D’Cruz, D.P. Diagnosis and classification of granulomatosis with polyangiitis (aka Wegener’s granulomatosis). J. Autoimmun. 2014, 48–49, 94–98.
- Ravin, K.A.; Loy, M. The Eosinophil in Infection. Clin. Rev. Allergy Immunol. 2016, 50, 214–227.
- Gangwar, R.S.; Levi-Schaffer, F. sCD48 is anti-inflammatory in Staphylococcus aureus Enterotoxin B-induced eosinophilic inflammation. Allergy 2016, 71, 829–839.
- Prince, L.R.; Graham, K.J.; Connolly, J.; Anwar, S.; Ridley, R.; Sabroe, I.; Foster, S.J.; Whyte, M.K. Staphylococcus aureus induces eosinophil cell death mediated by α-hemolysin. PLoS ONE 2012, 7, e31506.
- Gevaert, E.; Zhang, N.; Krysko, O.; Lan, F.; Holtappels, G.; De Ruyck, N.; Nauwynck, H.; Yousefi, S.; Simon, H.U.; Bachert, C. Extracellular eosinophilic traps in association with Staphylococcus aureus at the site of epithelial barrier defects in patients with severe airway inflammation. J. Allergy Clin. Immunol. 2017, 139, 1849–1860.e6.
- Kay, A.B. TH2-type cytokines in asthma. Ann. N. Y. Acad. Sci. 1996, 796, 1–8.
- Holgate, S.T. Innate and adaptive immune responses in asthma. Nat. Med. 2012, 18, 673–683.
- Le, K.Y.; Otto, M. Quorum-sensing regulation in staphylococci-an overview. Front. Microbiol. 2015, 6, 1174.
- Jiao, D.; Wong, C.K.; Qiu, H.N.; Dong, J.; Cai, Z.; Chu, M.; Hon, K.L.; Tsang, M.S.; Lam, C.W. NOD2 and TLR2 ligands trigger the activation of basophils and eosinophils by interacting with dermal fibroblasts in atopic dermatitis-like skin inflammation. Cell Mol. Immunol. 2016, 13, 535–550.
- Leyva-Castillo, J.M.; Das, M.; Kane, J.; Strakosha, M.; Singh, S.; Wong, D.S.H.; Horswill, A.R.; Karasuyama, H.; Brombacher, F.; Miller, L.S.; et al. Basophil-derived IL-4 promotes cutaneous Staphylococcus aureus infection. JCI Insight 2021, 6, e149953.
- Zhang, X.; Hu, X.; Rao, X. Apoptosis induced by Staphylococcus aureus toxins. Microbiol. Res. 2017, 205, 19–24.
- Kang, S.S.; Kim, S.K.; Baik, J.E.; Ko, E.B.; Ahn, K.B.; Yun, C.H.; Han, S.H. Staphylococcal LTA antagonizes the B cell-mitogenic potential of LPS. Sci. Rep. 2018, 8, 1496.