2. Mechanism of Biofilm Resistance
Antimicrobials (antibiotics, antivirals, antifungals, and antiparasitics) are compounds that kill microbes, stop their growth, and prevent or treat the infections in humans, animals, and plants. Antimicrobial resistance is the ability of microorganisms to survive against an antimicrobial drug at a higher concentration for a longer period, and is measured as minimum inhibitory concentration (MIC)
[6][23]. Biofilm resistance can be antibiotic resistance or antibiotic tolerance. Microorganisms develop mechanisms against antimicrobials either through acquisition of foreign genetic material coding for resistant determinants by horizontal gene transfer (HGT) in biofilm EPS, or through genetic mutation
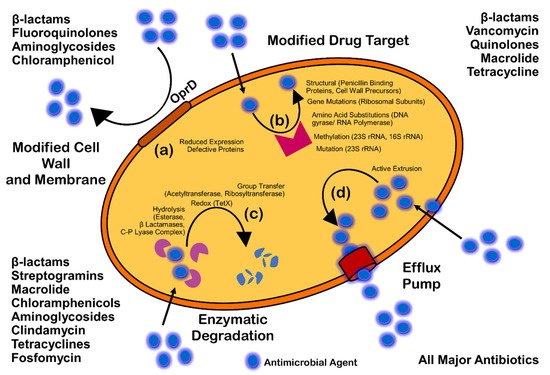
(Figure 3). Mechanisms of AMR are: prevention of access or reduced permeability of antimicrobials, mutational changes in antibiotic targets, modification of targets, and enzymatic degradation of the antimicrobials by hydrolysis or chemical change (
Figure 14). Antibiotic resistance (ABR) is a subdivision of AMR, as antibiotics are effective against the bacteria, but the bacteria become resistant to antibiotics.
Figure 14. Mechanism of antimicrobial resistance: cell wall modification (a); modification of drug target (b); enzymatic degradation or destruction of antimicrobials (c); and efflux pump (d).
Antimicrobial resistance can be intrinsic (naturally acquired) by genetic mutation, genes encoding inherent structural and functional traits of the molecular target, acquired extrinsically, or can be adaptive. Intrinsically, antibiotics such as vancomycin and daptomycin, which are active against Gram-positive bacteria, might not be effective against the Gram-negative bacteria due to distinct cell wall composition. On the other hand, acquired resistance involves genetic modification either through HGT or mutation. The bacteria can also adapt the capacity of resistance via gene expression and protein production rapidly in response to stress or other environmental conditions, and also in the presence of specific antibiotics. Thioredoxin A (
Ttrx A), thioredoxin reductase (
trxB), D-Ala-D-Ala carboxypeptidase,
DacA,
FabI, and
SapC are a few resistance genes which are responsible for the innate resistance to antibiotics such as fluoroquinolones, β-lactams, and aminoglycosides
[7][24].
However, tolerance is the ability of microorganisms to withstand or survive antibiotics higher than the inhibitory concentration for a period of time
[8][25]. Tolerance is an adaptive mechanism that reflects the change in cellular behavior from an active state to a dormant or inactive state
(Figure 3) for a transient period
[9][26]. A major rearrangement of energy production or few miscellaneous functions are witnessed during the tolerant state, and can be seen during poor growth or in the presence of few antibiotics or stress. Entrapment of antibiotics to the ECM without attachment to the target can also trigger tolerance, and results in dormancy or non-growth of bacterial cells. Persistence is a phenomenal form of tolerance, and persisters
(Figure 3) are the tolerant form of cells in the population that are capable of surviving antibiotics but can be killed with longer exposure
[10][27]. They can be either triggered persisters, induced in the presence of environmental stress or condition, or spontaneous persisters, converted to the form after a stochastic process. Persistence is also called heterotolerance
[11][28].
Biofilm-mediated resistance
(Figure 3) is a complex form of resistance that requires both the mechanisms of antibiotic resistance, as well as tolerance. Additionally, bacterial strains and species, antimicrobial agents, condition and developmental state of biofilm, and the growth condition of biofilm can highly affect the overall process
[8][25].
2.1. Prevention of Access or Reduced Penetration
The architecture, as well as composition, of ECM through gradients of dispersion can severely affect the penetration of antibiotics, the access to cells, and, finally, affect the efficacy of antibiotics. Diffusion of antibiotics also varies due to interaction with the ECM components
[12][29]. For example, extracellular DNA enhances the resistance of
Pseudomonas biofilm against aminoglycosides, but not against beta-lactams and fluoroquinolones
[13][14][15][30,31,32]. In the same way, eDNA enhances the resistance of
Staphylococcus epidermidis biofilm against glycopeptides. It has been seen that negatively charged eDNA binds to negatively charged glycopeptides (vancomycin) and aminoglycosides (tobramycin). It has also been demonstrated that the binding of vancomycin and eDNA is 100-fold higher than the vancomycin and D-Ala-D-Ala peptides in peptidoglycan precursors; this may result in the accumulation of eDNA in the ECM
[15][32]. The retention of eDNA in the ECM may result in a cation-limited environment through the chelation of Mg
2+ cations. The chelation of Mg
2+ can also initiate the AMR-linked two-component system of PhoPQ and PmrAB for
Psuedomonas aeruginosa and
S. enterica serovar Typhimurium
[14][16][31,33].
Additionally, the antibiotic modifying enzymes can be released and located in the ECM; these can also be used by other sensitive species of bacteria within a mixed species biofilm. For instance, beta lactamases released by
Moraxella catarrhalis protect the
S. pneumoniae and
H. influenza against amoxicillin and ampicillin, respectively
[17][18][34,35]. Therefore, the biofilm architecture can alter the diffusion of antibiotics, and also the exposure of cells.
2.2. Stress Responses and Nutritional Limitation
Physiological heterogeneity is characterized by the genetic and phenotypic expression, metabolic activity, and antibiotic tolerance between the cells within different areas of biofilm
[19][20][36,37]. The organization and architecture of biofilm generates gradients
(Figure 3) of dispersion of water, nutrients, pH, signaling molecules, and waste products. It is believed that cells near the surface of biofilm microcolony utilize most of the nutrient supplies and create a deprived area further down
[21][22][23][24][38,39,40,41]. Development of oxygen and nutritional depletion
(Figure 3) in microcolonies and lower niches can contribute to the development of diverse physiological states of aerobic, anaerobic, microaerobic, and fermentative conditions; also the development of persisters, slow growth, fast growth, and dormant cells
[19][25][17,36]. One such unique feature was observed by Yogesh and Anjali in 2021 (unpublished data), when MDR
Enterococcus faecalis strains were re-cultured directly from the biofilm stage, which were stored for an extended period of 16 to 18 months at −70 °C. They found that the re-cultured colonies could grow only after 60–72 h of incubation at 37 °C on nutrient or chromogenic UTI agar without any supplement. When examined by the investigators of this
study, the re-cultured bacterial strains were found to be resistant for the additional antibiotics. Cells in the oxygen deprived area
(Figure 3) can show reduced metabolic activities and may undergo dormancy; this is believed to be the reason for tolerance against antibiotics such as tobramycin and ciprofloxacin that target protein synthesis and DNA gyrase
[26][42].
It is well established that slow glowing cells are susceptible to colistin, which acts on the cell membrane
[27][43]. However, the presence of colistin-tolerant cells within oxygen rich areas was observed, suggesting disagreement with the connection of slow growth rate and development of antibiotic tolerance within biofilm
[28][29][44,45]. This fact was examined by Yogesh and Anjali in 2021 (unpublished data) when they found biofilm-producing
Enterococcus faecalis resistant to colistin. Despite the full thickness antibiotic penetration, visible cellular activities and protein synthesis have also been seen in oxygen rich areas
[30][31][46,47]. Through denitrification and fermentation,
P. aeruginosa can sustain the anaerobic conditions, and supplementation of nitrate or L-arginine can increase the metabolic activity within nutrient deprived areas, thereby increasing the susceptibility to ciprofloxacin and tobramycin
[26][42].
Stringent response (SR) and SOS response are adaptive mechanisms in response to stress and starvation of amino acids, iron, and carbon
[32][48]. The tolerance of
E. coli for cell wall inhibitor antibiotics such as carbapenems, penicillin, and cephalosporins is believed to be due to SR; this is also thought to be the case for cell division inhibitors such as norfloxacin and ofloxacin
[33][34][35][36][49,50,51,52]. DNA damage may induce the SOS response and can initiate antibiotic tolerance. DNA damage leads to the generation of single-stranded DNA; that may activate RecA, stimulate self-cleavage of the repressor LexA and result in the de-repression of SOS genes
[37][53]. The SOS response may lead to tolerance to antibiotics such as fluoroquinolones that can cause damage to DNA
[38][54]. The tolerance of
E. coli biofilm to fluoroquinolones has been observed due to the SOS response
[36][52].
2.3. Enzymatic Cell Wall Modification
The
dlt genes are crucial for
Enterococcus faecalis and
Staphylococcus aureus to form biofilm
[39][40][55,56]. This has been observed with the deletion of
S. aureus dltA and the subsequent reduction of resistance to vancomycin, and also the reduction of planktonic resistance of
E. faecalis to colistin and polymyxin B
[41][57]. The
dltABCD operon was a positive hit in a screening for biofilm-specific gentamicin tolerance genes in
Streptococcus mutans, a dental pathogen that can also cause infective endocarditis
[42][58]. The
dltABCD homologues are important for D-alanylation of teichoic acid in many Gram-positive species
[43][59]. Due to its inability to add D-alanine to the anionic teichoic acid molecules in the cell wall, the Δ
dltA mutant was more negatively charged than the wild-type
[42][58]. It is thought that the increased negative charge of the Δ
dltA strain promotes uptake of gentamicin, a positively charged aminoglycoside
[42][58].
2.4. Multispecies Interaction
The interaction between different species
(Figure 3) in biofilm can initiate antibiotic tolerance. For example, polymicrobial biofilms of
E. faecalis, Finogoldia magna, and
S. aureus were observed to be two-fold more resistant than the mono-species biofilm of
P. aeruginosa [44][60]. In the same way, in a dual species biofilm model,
M. catarhhalis released beta-lactamase, which protected
S. pneumoniae from amoxicillin
[17][45][34,61]. In the research by Ryan et al. (2008) on
P. aeruginosa and
Stenotrophomonas maltophilia dual-species biofilms, it was observed that the diffusible signal factor is an intercellular signaling molecule produced by
S. maltophilia, and is sensed by the two-component sensor BptS in
P. aeruginosa, leading to upregulation of the PmrA-regulated PA3552-3559 and PA4773-4775 genes
[46][62]. Furthermore, the interaction between fungi and bacteria in a multispecies biofilm has also been studied. The resistance of
Staphylococcus to vancomycin was increased in
C. albicans and
S. aureus biofilm due to the fungal matrix component beta-1,3-glucan, which is believed to act as a barrier against vancomycin
[47][48][63,64]. It was also noted that
C. albicans can increase the biofilm production of
P. aeruginosa through alcohol production
[49][65].
2.5. Mutation
Genomic mutation, even without any strong spontaneous selective stress or pressure, may lead to AMR. Mutation usually occurs at the rate of 10
−10 to 10
−9 per nucleotide per generation in most of the bacteria
[50][51][66,67]. Oxidative stress-causing agents that damage DNA can also accelerate the mutation rate. Although with a sublethal dose of a bactericidal, the build-up of ROS might be low but would be enough to induce synthesis of multidrug efflux pumps, mutagenesis, and promote resistance
[52][68]. The defect in
mutS,
mutL, and
uvrD genes can further promote the mutation frequency up to 100-fold due to failure of the DNA repair mechanism
[53][54][69,70]. An example of evolutionary mutation in microorganisms can be seen with hypermutators with the capability to acquire favorable mutations under selective stress that can lead to AMR
[55][71]. This particular phenotype has also been observed with
Pseudomonas biofilm, with resistance against rifampicin and ciprofloxacin
[56][72]. Apart from that the abovementioned phenotypic characteristics, hypermutations have also been reported in
S. aureus and
H. influenza isolated from cystic fibrosis infection but not in
Enterobacteriaceae isolated from acute urinary tract infection; which indicates hypermutability is favored in certain environments
[57][58][59][73,74,75].
Mutation in the
rspL gene, the gene that codes 16S rDNA and S12 ribosomal protein, affects the antibiotic targeting for aminoglycosides, whereas mutation in the
mexZ gene results in overproduction of the MexXY-OprM efflux system
[60][61][76,77]. Additionally, the mutations in the genes coding for the PmrAB two-component regulatory system, which regulates the addition of aminoarabinose to lipid A, has been associated with colistin resistance
[62][78]. Conversely, mutations in the promoter of the chromosomal
ampC gene that increase the plasmid copy number may result in increased production of β-lactamases
[63][64][79,80]. A large number of β-lactamase variants with point mutations in the gene, resulting in changes in the amino-acid sequence, may lead to the development of extended-spectrum β-lactamases (ESBLs) that also degrade first-, second-, and third-generation cephalosporins and/or became resistant to β-lactamase inhibitors
[65][81].
2.6. Efflux Pump
This is the movement of a drug from the intracellular to extracellular matrix without attachment to an intracellular target
(Figure 3 and Figure 4); therefore, this mechanism confers resistance to bacterial cells
[66][82]. Planktonic resistance in
P. aeruginosa to low concentration ofloxacin has been said to be due to the multiple multidrug efflux pumps, such asMaxAB-OprM
[67][83]. Another major multidrug efflux pump PA1875-1877 is believed to be a contributor to resistance in
P. aeruginosa biofilm
[68][84]. A two-fold to four-fold increase in susceptibility of biofilm to tobramycin, gentamicin, and ciprofloxacin was observed after the deletion of PA1875, PA1876, and PA1876; on the other hand, planktonic susceptibility was not affected much
[68][84]. In addition, the resistance of
P. aeruginosa biofilm to azithromycin was said to be due to the MexAB-OprM or MexCD-OprJ efflux pumps, whereas these efflux pumps are said to be required for the resistance against colistin in a metabolically active state
[28][69][44,85].
2.7. Quorum Sensing (QS)
QS is a population-density-dependent regulatory mechanism for interbacterial communication, which acts through signaling molecules named autoinducers. These autoinducers are recognized by the cell-surface receptors in order to induce gene transcription for virulence factors, surface proteins, transcriptional factors, and biofilm production
[70][71][86,87]. A biofilm formed by
P. aeruginosa lacking
lasR and
rhlR was observed as more susceptible to tobramycin than the wild-type biofilms
[72][88]. In the same way,
S. aureus deficient with QS-specific
agrD was observed as less resistant as compared to the wild-type
[73][89]. Moreover,
fsrA and
gelE mutants of
E. faecalis for QS and QS-controlled protease were seen with the impairment of biofilm formation in the presence of daptomycin, gentamicin, or linezolid
[74][90].