Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Catherine Yang and Version 1 by Satya Narayan.
Named as the guardian of the genome, p53 is a tumor suppressor that regulates cell function, often through many different mechanisms such as DNA repair, apoptosis, cell cycle arrest, senescence, metabolism, and autophagy. One of the genes that p53 activates is MDM2, which forms a negative feedback loop since MDM2 induces the degradation of p53. When p53 activity is inhibited, damaged cells do not undergo cell cycle arrest or apoptosis. As 50% of human cancers inactivate p53 by mutation, current research focuses on reactivating p53 by developing drugs that target the p53-MDM2 interaction, which includes the binding of MDM2 and phosphorylation of p53.
- p53
- Mdm2
- cancer
- small molecules
- chemotherapy
1. Introduction
P53 is also known as tumor suppressor protein p53 (TP53) and the guardian of the genome. It is a 393 amino acid transcription factor that regulates cell cycle progression. It can act as a transcriptional activator or repressor, which often leads to tumor suppression [1]. Depending on cellular stresses and cell cycle conditions, p53 can lead to various biological responses [2][3]. For example, if DNA damage is present, p53 is activated to transcribe target genes, such as Puma and Btg2, which induce DNA repair, apoptosis, cell cycle arrest, and senescence [4]. In addition, p53 is involved in metabolism and autophagy [5][6][7]. For instance, p53 prevents the metabolic reprogramming of cancer cells [7]. Moreover, autophagy, which can inhibit tumorigenesis by the engulfment and degradation of damaged cellular organelles, is promoted through the activation of p53-induced target genes, including Atg10 [1]. Thus, there are several ways by which p53 regulates cell function. As the guardian of the genome, p53 regulates cell proliferation and acts as a tumor suppressor. Since p53 is a highly studied molecule, there is a vast repository of literature on this topic. WThe researchers have tried to cite the most relevant references in this review. Our . The focus has been to describe some small molecules which have the potential for further development as therapeutic agents or that serve as leads for developing clinically viable agents targeting p53 and mouse double minute 2 homolog (MDM2) interaction.
MDM2 is a 491 amino acid regulator protein that consists of an N-terminal p53 binding domain, a nuclear localization signal (NLS), a NES, an acidic domain, a zinc-finger domain, and a C-terminal RING-finger domain. Similar to the binding of MDM2 at N-terminal activation domain of p53, the N-terminal p53-binding domain of MDM2 is an area that interacts with p53 (Figure 1). The NLS and the NES are essential for transporting MDM2 from the nucleus to the cytoplasm and vice versa. Within the acidic domain, residues are phosphorylated to induce the degradation of p53. Moreover, the C-terminal RING-finger domain functions as an E3 ubiquitin ligase that can induce the ubiquitination of p53 after p53-MDM2 interaction [8]. On the other hand, the zinc-finger domain, which is located next to the acidic domain, regulates the level of p53, as it interacts with ribosomal proteins that bind to the acidic domain and inhibits the degradation of p53 [9]. The MDM2 gene that expresses the protein includes 12 exons. The two promoters P1 and P2 containing p53 response elements (RE) are shown with red arrows. The P1 and P2 promoters regulate the expression of two MDM2 proteins, p90 and p76, respectively. P90 is a full-length and fully functional protein that can bind to p53 at the p53-binding domain. In contrast, P76, which is shorter than p90, acts as a negative inhibitor of p90 and activates p53 because it lacks the p53-binding domain [10][11]. Next to the two promoters, there are 10 additional exons, in which the first two are involved in the translation of p90 and p76. Using ATG as the first start codon, p90 and p76 are translated into exon 3 and 4, respectively. The p76-Mdm2 inhibits the ability of p90-Mdm2 to destabilize p53 [12].
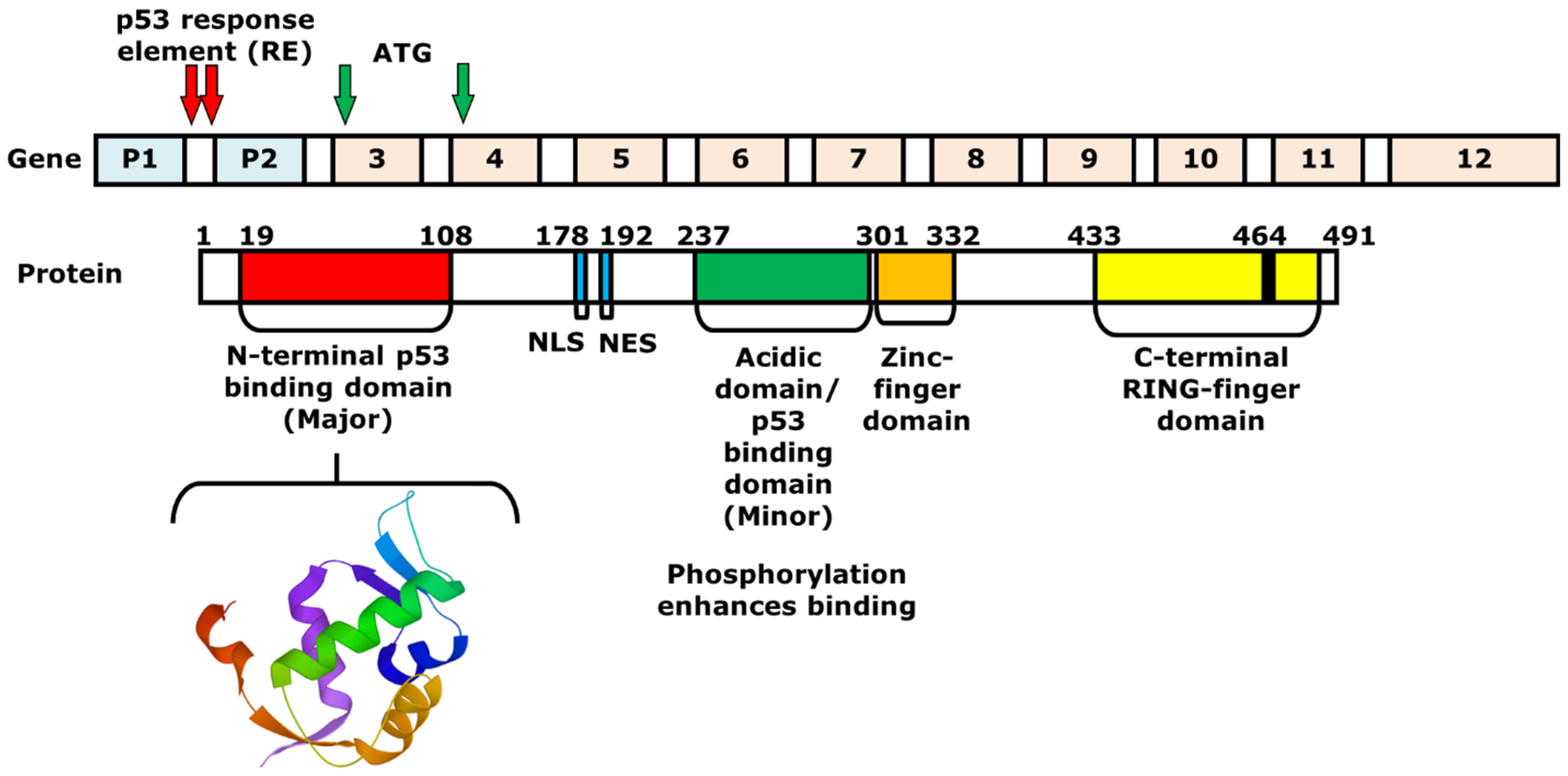
Figure 1. Structures of the MDM2 gene and protein. The 3D structure of a portion of MDM2 protein (17–111) is shown below, which consists of the N-terminal p53-binding domain (19–108). The MDM2 gene consists of two promoters and 10 additional exons. Between exons 1 and 2, or the first intron, there are two p53 response elements (REs), which are the two p53-binding sites. The REs are shown with red arrows. The full-length MDM2 protein p90 is produced by starting from ATG of exon 3, while the shorter protein, or p76, is synthesized with the start codon on exon 4.
2. Mechanism of p53 and MDM2-Binding
In normal cells or cells that are repaired, the concentration of p53 is low because of a regulator protein called the murine double minute 2 (MDM2). As mentioned above, the MDM2 gene is a negative regulator of p53, as it acts as a ubiquitin ligase [13][14]. It first physically interacts with p53 at TAD, which is amphipathic in nature. The binding pocket within the N-terminal p53-binding domain of MDM2 is hydrophobic in nature [15]. The p53 amino acid residues, including Phe19, Trp23, and Leu26, form the hydrophobic side of the amphipathic α-helix of the TAD. Although Phe19, Trp23, and Leu26 are not the only p53 residues that create the α-helix, these three amino acids play a key role in p53/MDM2 interactions [15]. Once the hydrophobic α-helix is formed, it contacts the hydrophobic binding pocket of MDM2, and thus, forms the p53/MDM2 complex via hydrogen bonding (Figure 2). During this interaction, Mdm2 induces the ubiquitination of p53 at the N-terminal activation domain, resulting in proteasomal degradation and inhibition of its transcriptional activity. One of p53 target genes is the cyclin-dependent kinase (CDK) inhibitor p21, which is a negative regulator of the cell cycle [16]. If the p53 is mutated, there will be no p21 activation, resulting the progression of the cell cycle even with damaged DNA [17]. As mentioned earlier, many of the human cancers inactivate p53 to allow for the continuation of cell cycle progression and cell survival. Thus, the p53-MDM2 interaction serves as an important focus area within cancer therapeutic studies. Current cancer therapeutics strive to increase levels of p53, by restoring the p53 function with the inhibition of its interaction with MDM2 or the degradation by MDM2. This strategy will prevent the survival and proliferation of tumor cells.
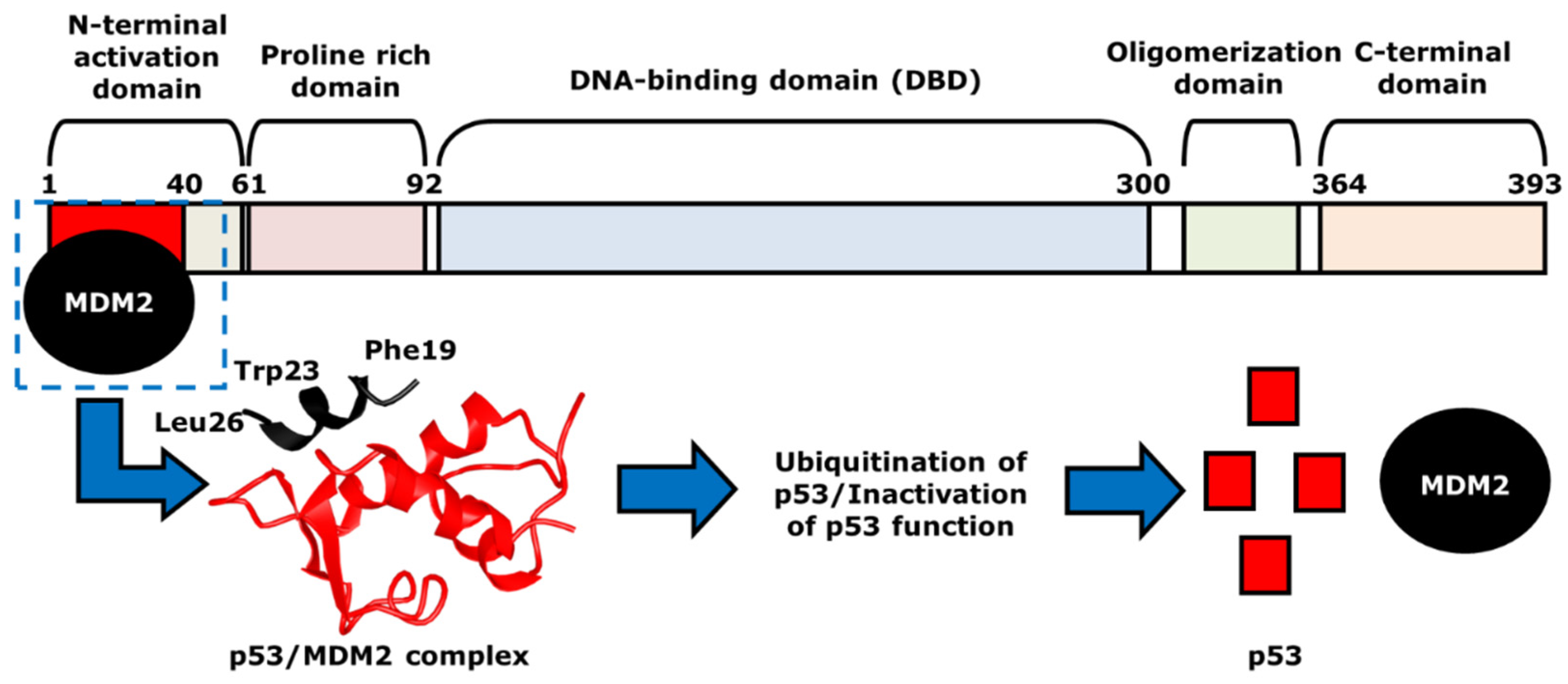
Figure 2. The mechanism of p53- and MDM2-binding. MDM2, indicated as black, binds to p53 at the N-terminal activation domain of p53, or the red area, to form p53/MDM2 complex. The two proteins are in direct contact via hydrogen bonding, with hydrophobic p53 residues Phe19, Trp23 and Leu26 that help such interaction. The structure of the p53/MDM2 complex, which is shown with the corresponding colors, black and red, leads to the ubiquitination and the proteasomal degradation of p53. This results in the inhibition of the transcriptional activity of p53.
3. The Role of p53 in Cancer Chemotherapy
P53 is activated when DNA damage is sensed by the protein kinases ataxia telangiectasia mutated (ATM) and ataxia telangiectasia and Rad3-related (ATR), which activate the checkpoint kinases Chk2 and Chk1, respectively [18][19]. The ATM-Chk2 pathway is activated by the double-strand DNA breaks (DSB), while the ATR-Chk1 pathway is triggered by single-strand DNA breaks [20]. Both Chk1 and Chk2 induce the phosphorylation of p53, specifically, at serine (Ser)-15 and Ser-20 of the N-terminal activation domain, and thus, stabilize and activate p53 function [21][22] As the levels of p53 increase, the activation of p53-dependent transcription of certain genes occurs, including p21 and MDM2 [23][24]. Similar to p53, p21 also acts as a tumor suppressor by regulating cell cycle and triggering apoptosis [25].
Under normal conditions, the complexes of CDK4/Cyclin D and CDK6/Cyclin D phosphorylate retinoblastoma tumor suppressor protein (Rb) and release the E2F transcription factor, which promotes the cell cycle to continue through the G1 phase into the S phase [26][27]. However, cells that express DNA damage increase p53-depnent expression of p21 causing cell cycle arrest at G1 by binding and inactivating CDK4-Cyclin D and CDK6/Cyclin D complexes. After the cell cycle arrest at G1 phase, the DNA damage can be repaired, and the cell cycle can progress through S phase. However, if the DNA damage cannot be fixed by the DNA repair machinery, there will be accumulation of DNA damage that can induce programmed cell death [28][29].
4. Strategies of Blocking the p53/MDM2 Interaction
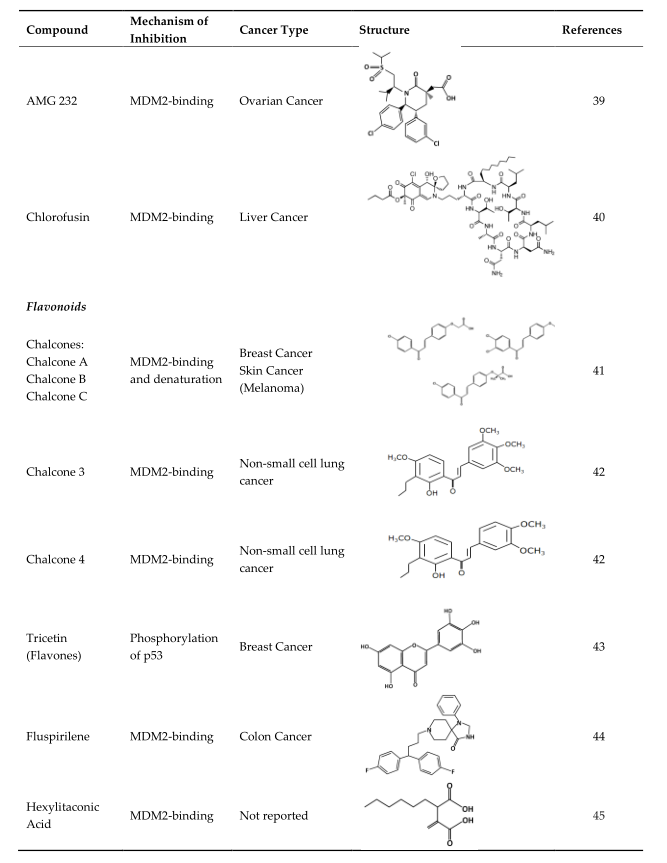
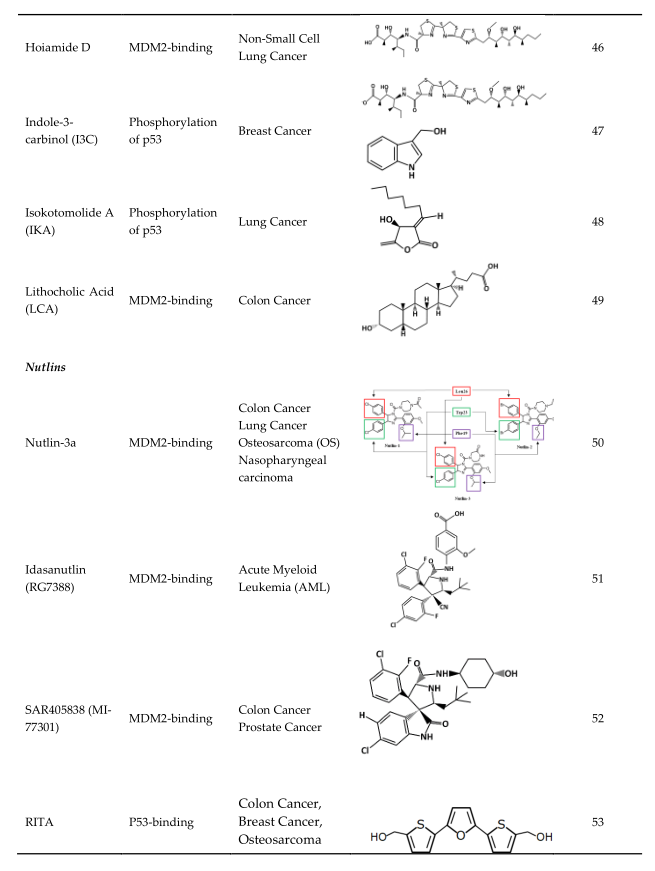
The blocking of the p53/MDM2 interaction can be achieved by the following two mechanisms: first, by inhibiting the p53 and MDM2-binding, and second, by increasing the phosphorylation of p53. In the MDM2-binding mechanism, several small molecules have been discovered that bind to MDM2 by mimicking the p53-binding pocket residues Phe19, Trp23, and Leu26 [30][31]. Prohibiting the binding of MDM2 to p53 allows for the restoration of p53 tumor suppressor function. On the other hand, instead of directing binding to MDM2, compounds can also prevent the interaction by inducing the phosphorylation of p53. Several studies showed that DNA double-strand breaks induce the phosphorylation of p53 at Ser15 by ATM or DNA-protein kinase (DNA-PK) and at Ser20 by Chk2 [32][33][34][35]. Both of these are part of MDM2 binding [36]. The phosphorylation of p53 at these residues abrogates binding with Mdm2, and thus it spares p53 from ubiquitination and proteasomal degradation [33]. Besides these two ways, there are several other mechanisms that can inhibit the interaction, such as the degradation of MDM2. However, this paperntry will focus on compounds that have MDM2 binding abilities or lead to the phosphorylation of p53 as mechanisms of action. The MDM2 residues that are critical for its interaction with p53 protein are G58, D68, V75, and C77 [37]. Some of the many existing compounds that target p53-MDM2 interaction and that may be used in future cancer treatments are summarized in Table 1 [38].
References
- Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53-mediated tumour suppression. Nature reviews Cancer. 2014;14(5):359-70. doi: 10.1038/nrc3711. PubMed PMID: 24739573; PubMed Central PMCID: PMC4049238.
- Vousden KH, Lu X. Live or let die: the cell's response to p53. Nature reviews Cancer. 2002;2(8):594-604. doi: 10.1038/nrc864. PubMed PMID: 12154352.
- Chen J. The Cell-Cycle Arrest and Apoptotic Functions of p53 in Tumor Initiation and Progression. Cold Spring Harb Perspect Med. 2016;6(3):a026104. doi: 10.1101/cshperspect.a026104. PubMed PMID: 26931810; PubMed Central PMCID: PMCPMC4772082.
- Vousden KH, Ryan KM. p53 and metabolism. Nature reviews Cancer. 2009;9(10):691-700. doi: 10.1038/nrc2715. PubMed PMID: 19759539.
- White E. Autophagy and p53. Cold Spring Harb Perspect Med. 2016;6(4):a026120. doi: 10.1101/cshperspect.a026120. PubMed PMID: 27037419; PubMed Central PMCID: PMCPMC4817743.
- Guo JY, White E. Autophagy, Metabolism, and Cancer. Cold Spring Harbor symposia on quantitative biology. 2016;81:73-8. doi: 10.1101/sqb.2016.81.030981. PubMed PMID: 28209717; PubMed Central PMCID: PMCPMC5521269.
- Fernandez-Fernandez MR, Sot B. The relevance of protein-protein interactions for p53 function: the CPE contribution. Protein engineering, design & selection : PEDS. 2011;24(1-2):41-51. doi: 10.1093/protein/gzq074. PubMed PMID: 20952436.
- Lindstrom MS, Jin A, Deisenroth C, White Wolf G, Zhang Y. Cancer-associated mutations in the MDM2 zinc finger domain disrupt ribosomal protein interaction and attenuate MDM2-induced p53 degradation. Molecular and cellular biology. 2007;27(3):1056-68. doi: 10.1128/MCB.01307-06. PubMed PMID: 17116689; PubMed Central PMCID: PMC1800693.
- Iwakuma T, Lozano G. MDM2, an introduction. Molecular cancer research : MCR. 2003;1(14):993-1000. PubMed PMID: 14707282.
- Loh SN. The missing zinc: p53 misfolding and cancer. Metallomics : integrated biometal science. 2010;2(7):442-9. doi: 10.1039/c003915b. PubMed PMID: 21072344.
- Perry ME, Mendrysa SM, Saucedo LJ, Tannous P, Holubar M. p76(MDM2) inhibits the ability of p90(MDM2) to destabilize p53. The Journal of biological chemistry. 2000;275(8):5733-8. doi: 10.1074/jbc.275.8.5733. PubMed PMID: 10681559.
- Takagi M, Absalon MJ, McLure KG, Kastan MB. Regulation of p53 translation and induction after DNA damage by ribosomal protein L26 and nucleolin. Cell. 2005;123(1):49-63. doi: 10.1016/j.cell.2005.07.034. PubMed PMID: 16213212.
- Manfredi JJ. Mdm2 and MdmX: Partners in p53 Destruction. Cancer research. 2021;81(7):1633-4. doi: 10.1158/0008-5472.CAN-21-0145. PubMed PMID: 34003788.
- Moll UM, Petrenko O. The MDM2-p53 interaction. Molecular cancer research : MCR. 2003;1(14):1001-8. PubMed PMID: 14707283.
- Johnson DG, Walker CL. Cyclins and cell cycle checkpoints. Annual review of pharmacology and toxicology. 1999;39:295-312. doi: 10.1146/annurev.pharmtox.39.1.295. PubMed PMID: 10331086.
- Cazzalini O, Scovassi AI, Savio M, Stivala LA, Prosperi E. Multiple roles of the cell cycle inhibitor p21(CDKN1A) in the DNA damage response. Mutation research. 2010;704(1-3):12-20. doi: 10.1016/j.mrrev.2010.01.009. PubMed PMID: 20096807.
- Marechal A, Zou L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harbor perspectives in biology. 2013;5(9). doi: 10.1101/cshperspect.a012716. PubMed PMID: 24003211; PubMed Central PMCID: PMCPMC3753707.
- Lanz MC, Dibitetto D, Smolka MB. DNA damage kinase signaling: checkpoint and repair at 30 years. The EMBO journal. 2019;38(18):e101801. doi: 10.15252/embj.2019101801. PubMed PMID: 31393028; PubMed Central PMCID: PMCPMC6745504.
- Smith J, Tho LM, Xu N, Gillespie DA. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Advances in cancer research. 2010;108:73-112. doi: 10.1016/B978-0-12-380888-2.00003-0. PubMed PMID: 21034966.
- Ou YH, Chung PH, Sun TP, Shieh SY. p53 C-terminal phosphorylation by CHK1 and CHK2 participates in the regulation of DNA-damage-induced C-terminal acetylation. Molecular biology of the cell. 2005;16(4):1684-95. doi: 10.1091/mbc.e04-08-0689. PubMed PMID: 15659650; PubMed Central PMCID: PMC1073652.
- Marei HE, Althani A, Afifi N, Hasan A, Caceci T, Pozzoli G, et al. p53 signaling in cancer progression and therapy. Cancer cell international. 2021;21(1):703. doi: 10.1186/s12935-021-02396-8. PubMed PMID: 34952583; PubMed Central PMCID: PMCPMC8709944.
- Hill R, Bodzak E, Blough MD, Lee PW. p53 Binding to the p21 promoter is dependent on the nature of DNA damage. Cell cycle. 2008;7(16):2535-43. doi: 10.4161/cc.7.16.6440. PubMed PMID: 18719376.
- Kachnic LA, Wu B, Wunsch H, Mekeel KL, DeFrank JS, Tang W, et al. The ability of p53 to activate downstream genes p21(WAF1/cip1) and MDM2, and cell cycle arrest following DNA damage is delayed and attenuated in scid cells deficient in the DNA-dependent protein kinase. The Journal of biological chemistry. 1999;274(19):13111-7. doi: 10.1074/jbc.274.19.13111. PubMed PMID: 10224064.
- Gartel AL. Is p21 an oncogene? Molecular cancer therapeutics. 2006;5(6):1385-6. doi: 10.1158/1535-7163.MCT-06-0163. PubMed PMID: 16818495.
- Narasimha AM, Kaulich M, Shapiro GS, Choi YJ, Sicinski P, Dowdy SF. Cyclin D activates the Rb tumor suppressor by mono-phosphorylation. Elife. 2014;3. doi: 10.7554/eLife.02872. PubMed PMID: 24876129; PubMed Central PMCID: PMC4076869.
- Day PJ, Cleasby A, Tickle IJ, O'Reilly M, Coyle JE, Holding FP, et al. Crystal structure of human CDK4 in complex with a D-type cyclin. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(11):4166-70. doi: 10.1073/pnas.0809645106. PubMed PMID: 19237565; PubMed Central PMCID: PMC2657441.
- Surova O, Zhivotovsky B. Various modes of cell death induced by DNA damage. Oncogene. 2013;32(33):3789-97. doi: 10.1038/onc.2012.556. PubMed PMID: 23208502.
- De Zio D, Cianfanelli V, Cecconi F. New insights into the link between DNA damage and apoptosis. Antioxidants & redox signaling. 2013;19(6):559-71. doi: 10.1089/ars.2012.4938. PubMed PMID: 23025416; PubMed Central PMCID: PMCPMC3717195.
- Shangary S, Wang S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction to reactivate p53 function: a novel approach for cancer therapy. Annual review of pharmacology and toxicology. 2009;49:223-41. doi: 10.1146/annurev.pharmtox.48.113006.094723. PubMed PMID: 18834305; PubMed Central PMCID: PMC2676449.
- Wang S, Zhao Y, Aguilar A, Bernard D, Yang CY. Targeting the MDM2-p53 Protein-Protein Interaction for New Cancer Therapy: Progress and Challenges. Cold Spring Harb Perspect Med. 2017;7(5). doi: 10.1101/cshperspect.a026245. PubMed PMID: 28270530; PubMed Central PMCID: PMCPMC5411684.
- Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;281(5383):1674-7. doi: 10.1126/science.281.5383.1674. PubMed PMID: 9733514.
- Shieh SY, Ikeda M, Taya Y, Prives C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell. 1997;91(3):325-34. doi: 10.1016/s0092-8674(00)80416-x. PubMed PMID: 9363941.
- Chehab NH, Malikzay A, Appel M, Halazonetis TD. Chk2/hCds1 functions as a DNA damage checkpoint in G(1) by stabilizing p53. Genes & development. 2000;14(3):278-88. PubMed PMID: 10673500; PubMed Central PMCID: PMCPMC316357.
- Shieh SY, Ahn J, Tamai K, Taya Y, Prives C. The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes & development. 2000;14(3):289-300. PubMed PMID: 10673501; PubMed Central PMCID: PMCPMC316358.
- Chen J. The Roles of MDM2 and MDMX Phosphorylation in Stress Signaling to p53. Genes Cancer. 2012;3(3-4):274-82. doi: 10.1177/1947601912454733. PubMed PMID: 23150760; PubMed Central PMCID: PMCPMC3494364.
- Freedman DA, Epstein CB, Roth JC, Levine AJ. A genetic approach to mapping the p53 binding site in the MDM2 protein. Molecular medicine. 1997;3(4):248-59. PubMed PMID: 9131587; PubMed Central PMCID: PMCPMC2230064.
- Beloglazkina A, Zyk N, Majouga A, Beloglazkina E. Recent Small-Molecule Inhibitors of the p53-MDM2 Protein-Protein Interaction. Molecules. 2020;25(5). doi: 10.3390/molecules25051211. PubMed PMID: 32156064; PubMed Central PMCID: PMCPMC7179467.
- Sahin I, Zhang S, Navaraj A, Zhou L, Dizon D, Safran H, et al. AMG-232 sensitizes high MDM2-expressing tumor cells to T-cell-mediated killing. Cell Death Discov. 2020;6:57. doi: 10.1038/s41420-020-0292-1. PubMed PMID: 32655895; PubMed Central PMCID: PMC7338458.
- Sun D, Li Z, Rew Y, Gribble M, Bartberger MD, Beck HP, et al. Discovery of AMG 232, a potent, selective, and orally bioavailable MDM2-p53 inhibitor in clinical development. Journal of medicinal chemistry. 2014;57(4):1454-72. doi: 10.1021/jm401753e. PubMed PMID: 24456472.
- Duncan SJ, Gruschow S, Williams DH, McNicholas C, Purewal R, Hajek M, et al. Isolation and structure elucidation of Chlorofusin, a novel p53-MDM2 antagonist from a Fusarium sp. Journal of the American Chemical Society. 2001;123(4):554-60. doi: 10.1021/ja002940p. PubMed PMID: 11456567.
- Balentine DA, Dwyer JT, Erdman JW, Jr., Ferruzzi MG, Gaine PC, Harnly JM, et al. Recommendations on reporting requirements for flavonoids in research. Am J Clin Nutr. 2015;101(6):1113-25. doi: 10.3945/ajcn.113.071274. PubMed PMID: 25854881.
- Pereira D LR, Palmeira A, Seca H, Soares J, Gomes S, Raimundo L, Maciel C, Pinto M, Sousa E, Vasconcelos MH, Saraiva L, Cidade H. Design and synthesis of new inhibitors of p53–MDM2 interaction with a chalcone scaffold. Arabian Journal of Chemistry. 2019;12(8):12. doi: doi.org/10.1016/j.arabjc.2016.04.015.
- Qin JJ, Li X, Hunt C, Wang W, Wang H, Zhang R. Natural products targeting the p53-MDM2 pathway and mutant p53: Recent advances and implications in cancer medicine. Genes & diseases. 2018;5(3):204-19. doi: 10.1016/j.gendis.2018.07.002. PubMed PMID: 30320185; PubMed Central PMCID: PMC6176154.
- Tichy A, Vavrova J, Pejchal J, Rezacova M. Ataxia-telangiectasia mutated kinase (ATM) as a central regulator of radiation-induced DNA damage response. Acta medica. 2010;53(1):13-7. doi: 10.14712/18059694.2016.57. PubMed PMID: 20608227.
- Nakahashi A, Miura N, Monde K, Tsukamoto S. Stereochemical studies of hexylitaconic acid, an inhibitor of p53-HDM2 interaction. Bioorganic & medicinal chemistry letters. 2009;19(11):3027-30. doi: 10.1016/j.bmcl.2009.04.057. PubMed PMID: 19414261.
- Bagal SK, Chapman ML, Marron BE, Prime R, Storer RI, Swain NA. Recent progress in sodium channel modulators for pain. Bioorganic & medicinal chemistry letters. 2014;24(16):3690-9. doi: 10.1016/j.bmcl.2014.06.038. PubMed PMID: 25060923.
- Katz E, Nisani S, Chamovitz DA. Indole-3-carbinol: a plant hormone combatting cancer. F1000Research. 2018;7. doi: 10.12688/f1000research.14127.1. PubMed PMID: 29904587; PubMed Central PMCID: PMC5989150.
- Brew CT, Aronchik I, Hsu JC, Sheen JH, Dickson RB, Bjeldanes LF, et al. Indole-3-carbinol activates the ATM signaling pathway independent of DNA damage to stabilize p53 and induce G1 arrest of human mammary epithelial cells. International journal of cancer Journal international du cancer. 2006;118(4):857-68. doi: 10.1002/ijc.21445. PubMed PMID: 16152627.
- Vogel SM, Bauer MR, Joerger AC, Wilcken R, Brandt T, Veprintsev DB, et al. Lithocholic acid is an endogenous inhibitor of MDM4 and MDM2. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(42):16906-10. doi: 10.1073/pnas.1215060109. PubMed PMID: 23035244; PubMed Central PMCID: PMC3479485.
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303(5659):844-8. doi: 10.1126/science.1092472. PubMed PMID: 14704432.
- Ding Q, Zhang Z, Liu JJ, Jiang N, Zhang J, Ross TM, et al. Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. Journal of medicinal chemistry. 2013;56(14):5979-83. doi: 10.1021/jm400487c. PubMed PMID: 23808545.
- Wang S, Sun W, Zhao Y, McEachern D, Meaux I, Barriere C, et al. SAR405838: an optimized inhibitor of MDM2-p53 interaction that induces complete and durable tumor regression. Cancer research. 2014;74(20):5855-65. doi: 10.1158/0008-5472.CAN-14-0799. PubMed PMID: 25145672; PubMed Central PMCID: PMC4247201.
- Issaeva N, Bozko P, Enge M, Protopopova M, Verhoef LG, Masucci M, et al. Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nature medicine. 2004;10(12):1321-8. doi: 10.1038/nm1146. PubMed PMID: 15558054.
More