The median overall survival of patients with metastatic breast cancer is only 2–3 years, and for patients with untreated liver metastasis, it is as short as 4–8 months. Improving the survival of women with breast cancer requires more effective anti-cancer strategies, especially for metastatic dis- ease. Nutrients can influence tumor microenvironments, and cancer metabolism can be manipulated via dietary modification to enhance anti-cancer strategies. Yet, there are no standard evidence-based recommendations for diet therapies before or during cancer treatment, and few studies provide definitive data that certain diets can mediate tumor progression or therapeutic effectiveness in human cancer. This review focuses on metastatic breast cancer, in particular liver metastatic forms, and recent studies on the impact of diets on disease progression and treatment.
- breast cancer liver metastasis
- western diet
- fasting-mimicking diet
1. Introduction
Data from the American Cancer Society estimate that there will be 1.9 million new cancer cases diagnosed and 608,570 cancer deaths in the US in 2021 [1]. For women in the US, breast cancer is the most common cancer (30% of all new cases), with an estimated 281,550 newly diagnosed cases and 43,600 deaths in 2021 [1]. In 2018, an estimated 3.7 million women were living with breast cancer in the US [2]. Furthermore, global breast cancer mortality is increasing substantially, especially in developing regions such as Latin America and the Caribbean, rising by an estimated 7 million deaths every five years [3]. These trends demonstrate a need for continued efforts to abate a serious public health concern.
One emerging approach to interveninge ion breast cancer outcomes is the use of targeted dietary interventions. Indeed, accumulating data indicates that practical clinical dietary interventions, such as the ketogenic diet, can improve the efficacy of anticancer therapy [4]. Thus, dietary approaches hold the potential to enhance therapeutic effectiveness and improve overall survival in breast cancer patients, thereby offering new promise for clinical practice that can change outcomes for a substantial number of patients worldwide.
Here, we review studies demonstrating how diet impacts disease progression and treatment in metastatic breast cancer, particularly metastases to the liver.
2. Breast Cancer Metastasis
Approximately 63% of breast cancer patients are diagnosed with local-stage breast cancer, 27% with the regional-stage disease, and 6% with distant (metastatic) disease [1]. In the US, an estimated >168,000 women were living with metastatic breast cancer in 2020 [5]. Al- though metastatic disease accounts for a small percentage of breast cancer cases, metastatic tumors are responsible for more than 90% of all cancer-related deaths [6]. Indeed, among breast cancer cases, the five-year survival rate for those with localized disease is more than 90%, but for those with metastases, the rate falls to just 28% [7]. Furthermore, the median survival of patients with metastatic disease at the time of diagnosis is approximately 18–24 months, and roughly 13% will survive 10 years [8]. About one-third of women diagnosed early with non-metastatic breast cancer will ultimately develop metastatic disease [9], which tends to develop resistance to therapies [10]. These phenomena underscore the increasing importance of developing therapies to prevent and treat metastatic disease and thus improve the overall survival of women with breast cancer [6].
The sites of distant metastasis among stage IV breast cancer patients include bone (68.8%), lung (16.0%), liver (13.3%), and brain (1.9%) [11]. Based on limited therapy options and dire disease outcomes for patients with liver metastasis, we will focus on liver metastatic ER+ breast cancer in this review. Important data on the impact of the location of metastases on patient survival come from the National Cancer Institute’s Surveillance, Epidemiology, and End Results (SEER), a network of tumor registries that include about 30% of the US population and harboring data from 1975 to 2017 [9]. Of the 2.4 million cancer patients within this database, 5.14% present with synchronous liver metastases (LM) [9[9],12[12]]. Half of all breast cancer patients develop LM, which often carries poor survival [13]—as low as 4–8 months if the disease is left untreated [14]. Surprisingly, metastatic breast cancer in the liver is observed more frequently in younger women (occurring in 34.2% of all patients < 50 years) than in older women (occurring in 8.9% of all patients ≥ 50 years) [15[15],16[16]]. In addition, patients with hormone receptor (HR)+/HER2+ breast cancer with LM have longer median survival than patients with HR+/HER2- and triple-negative breast cancer due to the introduction of HER2-targeted therapy [17[17],18[18]]. Thus, liver metastatic disease represents an important subgroup of breast cancer diagnoses that warrants focused efforts to improve outcomes.
3. Breast Cancer Liver Metastasis Diagnosis, Therapies, and Potential Treatments
Breast cancer LM may at first be asymptomatic, but possible symptoms include fatigue and weakness, pain or discomfort in the mid-section, weight loss or poor appetite, swelling in the legs, fever, and/or a yellow tint to the skin or whites of the eyes [19]. It is often identified by liver function tests that detect liver disease or damage [20]. Diagnosis may also be facilitated through imaging (MRI (magnetic resonance imaging), CT (computed tomography), PET (positron emission tomography), and PET/CT) or biopsy [21].
Most patients with breast cancer LM are treated with either systemic medications or local treatment [22]. Chemotherapy, hormonal therapies, or targeted therapies are common systemic treatments [23]. Chemotherapy involves the use of anti-cancer drugs to destroy or damage cancer cells [24]. Hormonal therapies use drugs such as tamoxifen, aromatase inhibitors, and fulvestrant to target estrogen and help shrink or slow the growth of HR+ metastatic breast cancer [25–27[25][26][27]]. Targeted therapies exploit specific characteristics of cancer cells to treat metastatic disease. Some common targeted therapeutics are everolimus, bevacizumab+paclitaxel, palbociclib, and ribociclib [28–33[28][29][30][31][32][33]]. Table 1 describes more current options such as potential oral selective estrogen receptor degraders or other pathway inhibitors. Local treatments for breast cancer LM include surgery, radiation therapy, and local chemotherapy. Surgery is most often used when the liver is the only site of metastasis and the symptoms are severe. Radiation therapies such as stereotactic body radiation therapy and Y-90 (Yttrium 90) radioembolization deliver or target radiation therapy directly to tumors in the liver [34[34],35[35]].
Endocrine therapies reduce breast cancer mortality and relieve symptoms, but some persistent tumor cells frequently develop resistance in the metastatic and adjuvant setting [36–38[36][37][38]]. Liver metastatic estrogen receptor α (ERα)-positive breast cancer is currently incurable [39]. Some potential small molecule therapies show good tumor responses in metastatic breast cancers. Axl kinase is associated with aggressive migratory behavior in tumors in a mouse model, and a combination of R428, a selective small molecule Axl inhibitor, with cisplatin positively reinforces both agents to block liver micro-metastases [40][40]. VERU-111 acts by depolymerizing microtubules, often leading to cell apoptosis due to the inability to complete mitosis, and is highly effective, especially against fibrous tumors and metastases [41]. Recent evidence indicates that ErSO, a small molecule activator of a stress response mechanism that stimulates the anticipatory unfolded protein response (a-UPR), can eradicate most lung, bone, and liver metastases in orthotopic cell line xenograft and patient-derived xenograft (PDX) mouse models [39].
| Therapy | Administration | Target | Combination | Status | Year |
|---|---|---|---|---|---|
| Everolimus [42] | Oral | mTOR | Not noted | FDA approved | 2020 |
| Alpelisib [28[28],42[42]] | Oral | PI3K-alpha | Combination with fulvestrantor letrozole |
FDA approved | 2020 |
| Elacestrant [43] | Oral | Estrogen receptor | Low-fat diet combination | Phase Ib | 2020 |
| Giredestrant [44] | Oral | Estrogen receptor | Not noted | Phase III | 2021 |
| AZD9833 [45] | Oral | Estrogen receptor | Not noted | Phase I | 2020 |
4. Link between Diets and Metastatic Breast Cancer
Dietary factors account for about 30% of cancer cases; thus, diet is one of the most modifiable causes of cancer [46]. High consumption of red meat, animal fats, and re- fined carbohydrates is associated with increased risk and severity of diseases such as breast cancer [47–51[47][48][49][50][51]]. Furthermore, postmenopausal women have an increased risk of developing obesity-related breast cancer due to Western diets that promote weight gain, fat redistribution, dyslipidemia, hypertension, and insulin resistance, all of which are important in the recognition of metabolic syndrome [52–54[52][53][54]]. For overweight or obese women, postmenopausal estrogen receptor-positive (ER+) and progesterone receptor-positive (HR+) breast cancer risks are about 1.5–2 times that of women with normal body weights [55[55],56[56]]. This could be due to higher levels of estrogen produced by extra fat tissues in postmenopausal women and/or other mechanisms such as elevated levels of insulin [57[57],58[58]]. Many studies have shown that weight gain also increases the risk of breast cancer in postmenopausal women compared with normal-weight women [59–63[59][60][61][62][63]].
4.1. Western Diet
The Western diet is rich in fat and sugar, involving a high intake of saturated fats and sucrose and a low intake of fiber [64]. It plays a role in inflammatory disease and negatively affects both the immune system and gut microbiota [65]. Western diets are strongly associated with obesity and other metabolic effects such as weight gain and are often blamed for “the obesity epidemic”, as well as rising incidences of type 1 and type 2 diabetes [66].
Glucose is central to the Western diet, so it is important to understand how cancer cells behave on a primary glucose energy source. (Figure 1). The Western diet directly promotes tumor cell proliferation via mechanisms involving the insulin/insulin-like growth factor 1 (IGF1)/phosphoinositide 3-kinase (PI3K) signaling pathway [67]. During regular cell function, glucose stimulates pancreatic β cells to release insulin, allowing glucose to enter cells to be used as a fuel source [68]. The high intake of carbohydrates and glucose stimulates the pancreas to increasingly secrete more insulin, which promotes the interaction of growth hormone receptors and growth hormones [69]. This elevates the levels of free IGF-1 released from the liver, which are associated with cell growth and proliferation and can harm cancer patients [70]. IGF-1 stimulates phosphorylation and activation of the serine/threonine kinase Akt via PI3K signaling. Akt then activates the mammalian target of rapamycin (mTOR) and induces aerobic glycolysis by c-Myc and hypoxia-inducible factor (HIF)-1α. Insulin also stimulates interleukin 6 (IL-6) and tumor necrosis factor α (TNF-α) release [71–73[71][72][73]].
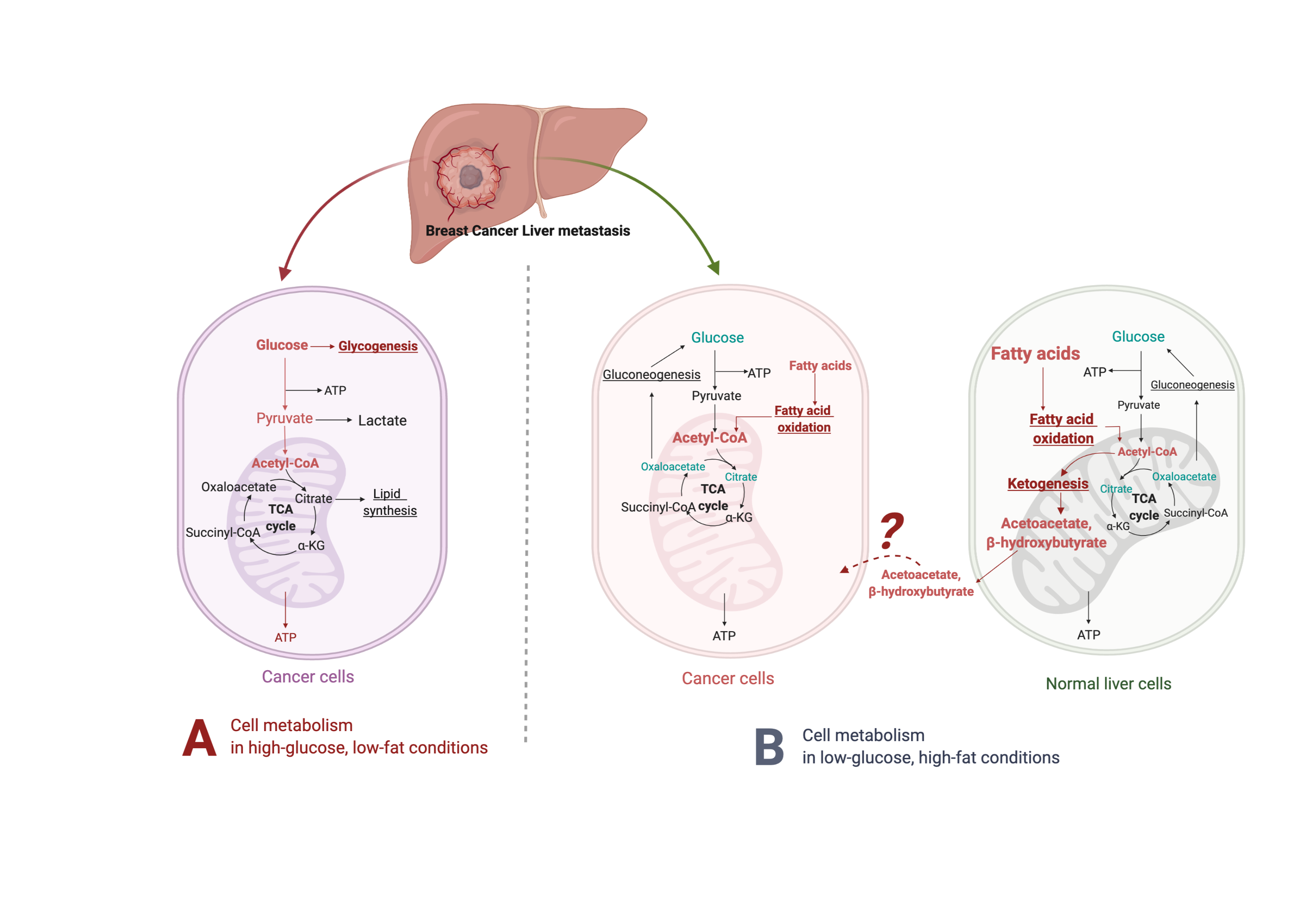
Figure 1. Suggested liver metastatic cancer metabolism in normal diet or fasting-mimicking diet. In the presence of glucose, the breast cancer cells metastasized to the liver mainly go through glycolysis to produce pyruvate, which is converted to acetyl-CoA via oxidative decarboxylation. Acetyl-CoA enters the tricarboxylic acid cycle (TCA cycle) and generates adenosine-3-phosphate (ATP) for cell survival and proliferation. Excessive glucose can be stored as glycogen for further usage. When glucose is limited (under a fasting-mimicking diet), the cancer cells switch to acquire ATP by fatty acid oxidation. The fatty acid oxidation is also highly activated in peripheral hepatocytes, where abundant acetyl-CoA feeds into ketogenesis and produces a large amount of acetoacetate and β- hydroxybutyrate. The liver does not use ketone bodies for energy because it lacks the necessary enzyme thiophorase (beta ketoacyl-CoA transferase). The unclear part is how these released ketone bodies function on breast cancer cells.
There is evidence that deregulated glucose signaling influences cancer. Overexpression of glucose transporters 1 and 3 occurs in many aggressive tumors, and this correlates with elevated glucose uptake [74[74],75[75]]. Furthermore, reducing glucose concentrations significantly decreases the proliferation of MCF-7 and T47D breast cancer cells and MCF-10A breast epithelial cells [76]. High glucose (25 mM) levels significantly abrogate the therapeutic effects of metformin on triple-negative breast cancer cell proliferation, death, and cell cycle arrest and contributed to metastatic progression and the development of resistance to chemotherapy/radiotherapy [77]. Additionally, mice with breast cancer liver metastasis fed sugar-rich diets had a high metastatic burden, while mice fed high-fat/low-sugar diets had a low tumor burden despite obesity [78]. In other mice studies, hepatocellular carcinoma tumor burden positively correlated with hepatic fat accumulation and insulin and liver IL-6 levels and inversely correlated with adiponectin levels [79[79],80[80]]. These results indicate that dietary sugar intake may stimulate liver tumor growth.
Analysis of metabolic pathway components in mice indicates that reduced extracellular glucose can stimulate coactivator-associated arginine methyltransferase 1 (CARM1) to methylate GAPDH at R234, decreasing its likelihood of associating with its coenzyme NAD+. This inhibits the enzymatic activity of GAPDH and represses glycolysis to delay liver cancer cell growth, as cancer cells depend on glycolysis for proliferation [81]. Numerous other studies found that cancer cells become more dependent on blood glucose due to the demand for rapid cell growth, while others indicate that glucose may directly or indirectly affect tumor cell proliferation [77,82–84[77][82][83][84]]. Notably, there is a higher incidence of breast cancer among diabetic and obese populations, contributing to the theory that a low-carbohydrate diet may limit or prevent tumor growth [85]. Emerging evidence supports the role of dietary interventions to counteract this putative effect.
4.2. Fasting-Mimicking Diet
The fasting-mimicking diet is low in calories from sugars, and protein but high in unsaturated fats. This diet is widely studied in relation to disease prevention and treatment, and although it has low levels of toxicity, it may have limits in terms of diet adherence and preexisting nutritional deficiencies [86]. Unlike the Western diet, low-carbohydrate diets slow cancer by inhibiting insulin/IGF and downstream intracellular signaling pathways, specifically PI3K/Akt/mTOR. Indeed, fasting prevents a Warburg shift, curbs glycolysis, and impedes AKT/mTOR signaling to improve the therapeutic response of Sorafenib-resistant hepatocellular carcinoma via p53-dependent metabolic synergism [82]. Increased AMP-activated protein kinase (AMPK) levels, stimulated by adenosine monophosphate, inhibit aerobic glycolysis and suppress proliferation, migration, and invasion of tumor cells [87].
Fluorodeoxyglucose-positron emission tomography (FDG-PET) demonstrates that most human cancer cells have a higher demand for glucose than surrounding non-cancer cells [88[88],89[89]]. Metastatic cancer cells typically resemble cells of primary cancer, but they can also be influenced by the milieu of the organs they colonize. Metabolic reprogramming happens after cells metastasize and colonize the liver. Liver cancer cells, like most other cancers, perform metabolic rewiring to increase their energy metabolism, becoming depen- dent on glucose or fructose as energy sources to fuel high rates of glycolysis or fructolysis, respectively [75,90–92[75][90][91][92]], resulting in the use of the pentose phosphate pathway and glycoly- sis to generate NADPH and pyruvate [93–95[93][94][95]] (Figure 1, left panel). For instance, dietary fructose provides fuel for major pathways of central carbon metabolism during tumor cell proliferation by activating the enzyme aldolase B (ALDOB) or its upstream regulator GATA6 in colon cancer liver metastasis [92]. Cancer cells become dependent on adenosine triphosphate (ATP) produced by the less efficient process of glycolysis [96]. Furthermore, tumor cells have more mitochondrial DNA mutations than normal cells, producing an increased number of reactive oxygen species (ROS) during respiration [97]. These cells are less capable of producing NADPH because gluconeogenesis cannot be performed to form the glucose-6-phosphate (G-6-P) necessary to enter the pentose phosphate pathway [98].
When glucose is limited, the body produces an alternative form of energy for its cells. Under the fasting-mimicking diet, cancer cells are forced to use mitochondrial oxidative metabolism, which causes metabolic oxidative stress as well as the production of ketones for energy instead (Figure 1, right panel). Ketone bodies produced by the liver benefit normal cells but not cancer cells [99[99],100[100]]. As part of the Warburg effect, lactate is produced in excess, which compensates for dysfunctional mitochondrial oxidative phosphorylation [74[74],101[101],102[102]]. High-fat, low-carbohydrate diets such as the fasting-mimicking diet are notable due to their ability to restrict the availability of glucose and limit a Warburg-type metabolism [103], further supporting that these diets have the potential to prevent or reverse tumor growth.
These series of events mean tumor cells are dependent on glucose, and this depen- dency can be exploited with the fasting-mimicking diet to selectively starve tumors by providing fat and protein that the tumor cells cannot use [104]. Several animal studies of various cancer types showed that the fasting-mimicking diet effectively limits tumor growth by itself or in combination with other therapies without causing the rebound hyper- glycemia and hyperinsulinemia [100,101,105–113[100][101][105][106][107][108][109][110][111][112][113]]. Metastatic TNBC patients with lower glycemia survive longer compared with those with higher glycemia. FMD reduces TNBC cancer stem cells (CSCs) and delays tumor progression [114]. Synergistic anti-neoplastic effects of the metformin/hypoglycemia combination were regulated by PP2A-GSK3β- MCL-1 axis, leading to a decline in the pro-survival protein MCL-1 and reduction in tumor growth in in vitro and in vivo metastatic melanomas models [115]. Furthermore, the very low carbohydrate diet reduces tumor incidence in a spontaneous mouse model of breast cancer [101]. In the metastatic 4T1 mouse mammary tumor model, combining a low-carbohydrate/high-protein diet and a cyclooxygenase-2 inhibitor significantly lowers the levels of breast cancer lung metastasis [108]. A low-carbohydrate diet also suppresses prostate cancer tumor growth in mice compared to a Western diet, which increases serum insulin, blood glucose, and tumor tissue insulin receptor levels [107]. Finally, a fasting-mimicking diet synergizes with classical chemotherapy to treat metastatic murine pancreas cancer in preclinical models by decreasing tumor glucose and glycolytic intermediates, increasing β-hydroxybutyrate, and boosting reactive oxygen species [112].
4.3. β-Hydroxybutyrate Paradox
On a high-fat/low-carbohydrate diet, a process called ketogenesis produces ketone bodies in the liver [103[103],116[116]] through the production of acetyl-coenzyme A (CoA) [117]. When the supply of liver carbohydrates is low, acetyl-CoA is broken down to acetoacetate (AcAc) and then further reduced to β-hydroxybutyrate (βHB), one of the most abun- dant and principle ketone bodies [116[116],118[118]]. Though βHB is derived in the liver from the β-oxidation of free fatty acids (FFAs), the liver does not use ketone bodies for energy because it lacks the necessary enzyme thiophorase (beta ketoacyl-CoA transferase) [119] (Figure 1, right panel). In most humans, the plasma βHB concentration is typically at least 2 mM on a low-carbohydrate/high-fat diet [120]. βHB, a major metabolite of the fasting-mimicking diet, helps regulate many metabolic diseases due to its ability to control signaling events [116], e.g., the PI3K/Akt/mTOR pathways [110[110],121[121],122[122]]. A fasting-mimicking diet enhances the anti-cancer efficacy of the endocrine therapeutics including tamoxifen and fulvestrant and delays endocrine resistance by lowering circulating IGF1, insulin, and leptin and by inhibiting AKT–mTOR signaling via upregulation of EGR1 and PTEN in mouse models of hormone-receptor-positive breast cancer [110]. Vernieri, Claudio, et al. reported the FMD first-in-human clinical trial (NCT03340935) in patients with different tumor types (including 56 breast cancer patients, 26 luminal, 19 TNBC, and 11 HER+) and treated with concomitant antitumor therapies. The FMD favorably modulates systemic and intratumor immunity and activates several antitumor immune programs by significantly reducing plasma glucose concentration, serum insulin, and serum IGF1 [123]. Some of its other key molecular targets include the NLRP3 inflammasome, RNA-binding proteins, and G protein-coupled receptors [124].
βHB has anti-inflammatory properties [122[122],125[125]] and is characterized as an epigenetic modifier that produces anti-cancer effects by modifying chromatin and inhibiting histone deacetylases [126[126],127[127]]. However, some studies link βHB to tumor progression, metastasis, and clinical failure [128–131[128][129][130][131]]. These inverse effects gave rise to the “β-hydroxybutyrate paradox” theory [131].
Metastatic cancer models indicate that exogenous ketones have direct cytotoxic effects on the viability and survival of tumors [87]. For example, βHB effectively inhibits S2-013 cells, a cloned subline derived from a liver metastasis, with consequent metabolic reprogramming, of a human pancreatic tumor line (SUIT-2) [132]. Maldonado et al. demonstrated that low-carbohydrate diet-induced glucose deprivation is a potential strategy to enhance breast cancer treatment—very high βHB levels (25 mM) do not stimulate breast cancer cell proliferation, suggesting that breast cancer cells cannot use βHB as fuel to proliferate [76]. In an in vivo study of mice implanted with VM-M3 tumors, dietary ketone supplementation (either 1,3-butanediol or a ketone ester, which are metabolized to the ketone bodies βHB and acetoacetate) prolonged survival and reduced tumor burden in mice with metastatic cancer. In addition, supplementation lowers blood glucose, elevates blood ketones, and decreases overall body weight [87]. In another study, βHB enhances cisplatin-induced apoptosis via the histone deacetylase (HDAC)3/6 inhibition/survival axis in hepatocellular carcinoma [133]. Furthermore, clinical trials at the University of Würzburg tested low-carbohydrate/high-fat diets in 16 patients with advanced/metastatic solid malignant tumors and found that 3 months of ketogenic diet therapy resulted in a stable physical condition, lower body mass index, a somewhat better quality of life, and/or slowed tumor growth [134].
The β-hydroxybutyrate paradox indicates that βHB’s effect on cancer growth de- pends on the tumor’s energetic phenotype. Cells with an “oxidative phosphorylation phenotype” use βHB as an additional energy source whenever it is available, while cells with a “glycolytic, Warburg-like phenotype” cannot metabolize βHB, causing it to accumulate within the cell and inhibit tumor growth through cell signaling and epigenetic mechanisms [131[131],135[135]]. An in vitro study determined that βHB can change the energetic phenotype of breast cancer cells but not their glucose consumption and production of lactate [135]. Furthermore, in a spontaneous mouse mammary tumor model, βHB at low concentration (<1 mM) increased tumor growth by acting as an oxidative energy source rather than as an epigenetic factor [131].
In addition to the β-hydroxybutyrate paradox, there is a paradox surrounding the common ketone body butyrate. The butyrate paradox suggests that, like βHB, butyrate can mediate histone acetylation and inhibit cell proliferation in cells following the War- burg effect and preferentially using glucose [131,136–138[131][136][137][138]]. However, in cancer cells that do not follow the Warburg effect and oxidize butyrate as fuel, butyrate fails to reach inhibitory concentrations and can stimulate tumor growth [131]. An in vitro study showed that sodium butyrate (NaBu), an HDAC inhibitor, inhibits breast cancer cell growth in a time- and dose-dependent manner. This possible anti-cancer effect is due to NaBu eliciting apoptosis through elevated levels of ROS, increased caspase activity, and reduced mitochondrial membrane potential [139]. Luo et al. also demonstrated that NaBu induces autophagy in colorectal cancer cells through phosphorylated liver kinase B1 (LKB1)/AMPK signaling [138].
Overall, there are still many controversial opinions about high-fat/low-glucose diets, and the scientific community has not reached a consensus on their benefits or detriments. However, a fasting-mimicking diet can reduce the toxic effects of chemotherapy and enhance therapeutic efficacy beyond chemotherapy alone [140[140],141[141]]. When used alongside chemotherapy, the fasting-mimicking diet delays breast cancer and melanoma progression in mice by reducing HO-1 to sensitize tumors to chemotherapy [142[142]]. When applied to individuals with HER2-early breast cancer during chemotherapy, the fasting-mimicking diet increased tumor cell death and significantly slowed chemotherapy-induced DNA damage in T-lymphocytes [140[140],141[141]]. Both animal studies and clinical trials are ongoing to better understand the mechanism of the fasting-mimicking diet and βHB-altered tumor microenvironments in specific cancer types.
5. Conclusions and Future Perspective
Globally, breast cancer is the most prevalent cancer in women, and metastatic disease is highly predictive of shortened survival. Improving the general survival of women with metastatic breast cancer requires more effective anti-cancer strategies in combination with current therapies or medications. Nutrition plays an important role before/during/after cancer treatment. While dietary modifications mostly have positive effects in the context of specific cancers, it is critical to optimize future investigations on metabolic therapies to understand how dietary factors and pharmacotherapies influence carcinogenesis based on tumor- and patient-related characteristics. To further improve the effects of the fasting-mimicking diet on the quality of life or cancer progression, more clinical studies are needed, as the safety and efficacy of the fasting-mimicking diet strongly depend on the tumor variety and its genotype. Understanding diet-associated molecular mechanisms involved in therapy resistance, especially the “β-hydroxybutyrate paradox theory”, will help reduce mortality and morbidity associated with metastatic breast cancer and prolong the quality-adjusted life expectancy of cancer survivors.[1] variety and its genotype. Understanding diet-associated molecular mechanisms involved in therapy resistance, especially the “β-hydroxybutyrate paradox theory”, will help reduce mortality and morbidity associated with metastatic breast cancer and prolong the quality-adjusted life expectancy of cancer survivors.
References
- 1. Tarver, T. Breast Cancer Facts & Figures 2021; American Cancer Society: Atlanta, GA, USA, 2021.2. National Cancer Institute. Cancer Stat Facts: Female Breast Cancer; National Cancer Institute: Bethesda, MD, USA, 2020.3. Azamjah, N.; Soltan-Zadeh, Y.; Zayeri, F. Global Trend of Breast Cancer Mortality Rate: A 25-Year Study. Asian Pac. J. Cancer Prev. 2019, 20, 2015–2020. [CrossRef] [PubMed]4. Lévesque, S.; Pol, J.G.; Ferrere, G.; Galluzzi, L.; Zitvogel, L.; Kroemer, G. Trial watch: Dietary interventions for cancer therapy. OncoImmunology 2019, 8, e1591878. [CrossRef]5. Mariotto, A.B.; Etzioni, R.; Hurlbert, M.; Penberthy, L.; Mayer, M. Estimation of the Number of Women Living with Metastatic Breast Cancer in the United States. Cancer Epidemiol. Prev. Biomark. 2017, 26, 809–815. [CrossRef] [PubMed]6. Gupta, G.P.; Massagué, J. Cancer metastasis: Building a framework. Cell 2006, 127, 679–695.[CrossRef] [PubMed]7. Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [CrossRef] [PubMed]8. Eng, L.G.; Dawood, S.; Sopik, V.; Haaland, B.; Tan, P.S.; Bhoo-Pathy, N.; Warner, E.; Iqbal, J.; Narod, S.A.; Dent, R. Ten-year survival in women with primary stage IV breast cancer. Breast Cancer Res. Treat. 2016, 160, 145–152. [CrossRef] [PubMed]9. Howlader, N.; Noone, A.M.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; et al. SEER Cancer Statistics Review, 1975–2017; National Cancer Institute: Bethesda, MD, USA, 2020. Available online: https://seer.cancer.gov/csr/1975_2017/ (accessed on 5 April 2022).10. Soni, D.A.; Ren, Z.; Hameed, O.; Chanda, D.; Morgan, C.J.; Siegal, G.P.; Wei, S. Breast Cancer Subtypes Predispose the Site of Distant Metastases. Am. J. Clin. Pathol. 2015, 143, 471–478. [CrossRef]11. Gong, Y.; Liu, Y.-R.; Ji, P.; Hu, X.; Shao, Z.-M. Impact of molecular subtypes on metastatic breast cancer patients: A SEER population-based study. Sci. Rep. 2017, 7, 45411. [CrossRef]12. Horn, S.R.; Stoltzfus, K.C.; Lehrer, E.J.; Dawson, L.A.; Tchelebi, L.; Gusani, N.J.; Sharma, N.K.; Chen, H.; Trifiletti, D.M.; Zaorsky,N.G. Epidemiology of liver metastases. Cancer Epidemiol. 2020, 67, 101760. [CrossRef] [PubMed]13. Rashid, N.S.; Grible, J.M.; Clevenger, C.V.; Harrell, J.C. Breast cancer liver metastasis: Current and future treatment approaches. Clin. Exp. Metastasis 2021, 38, 263–277. [CrossRef] [PubMed]14. Adam, R.; Aloia, T.; Krissat, J.; Bralet, M.P.; Paule, B.; Giacchetti, S.; Delvart, V.; Azoulay, D.; Bishmuth, H.; Castaing, D. Is liver resection justified for patients with hepatic metastases from breast cancer? Ann. Surg. 2006, 244, 897. [CrossRef] [PubMed]15. De Ridder, J.; De Wilt, J.H.W.; Simmer, F.; Overbeek, L.; Lemmens, V.; Nagtegaal, I. Incidence and origin of histologically confirmed liver metastases: An explorative case-study of 23,154 patients. Oncotarget 2016, 7, 55368–55376. [CrossRef] [PubMed]16. Cummings, M.C.; Simpson, P.T.; Reid, L.E.; Jayanthan, J.; Skerman, J.; Song, S.; Reed, A.E.M.; Kutasovic, J.R.; Morey, A.L.; Marquart, L.; et al. Metastatic progression of breast cancer: Insights from 50 years of autopsies. J. Pathol. 2014, 232, 23–31.[CrossRef] [PubMed]17. Ji, L.; Cheng, L.; Zhu, X.; Gao, Y.; Fan, L.; Wang, Z. Risk and prognostic factors of breast cancer with liver metastases. BMC Cancer 2021, 21, 1–15. [CrossRef]18. Xie, J.Z.; Xu, A. Population-Based Study on Liver Metastases in Women with Newly Diagnosed Breast Cancer. Cancer Epidemiol Biomark. Prev. 2019, 28, 283–292. [CrossRef]19. Diamond, J.R.; Finlayson, C.A.; Borges, V.F. Hepatic complications of breast cancer. Lancet Oncol. 2009, 10, 615–621. [CrossRef]20. Patanaphan, V.; Salazar, O.M.; Risco, R. Breast cancer: Metastatic patterns and their prognosis. South. Med. J. 1988, 81, 1109–1112. [CrossRef]21. Cao, R.; Wang, L.-P. Serological Diagnosis of Liver Metastasis in Patients with Breast Cancer. Cancer Biol. Med. 2012, 9, 57–62. [CrossRef]22. Bale, R.; Putzer, D.; Schullian, P. Local Treatment of Breast Cancer Liver Metastasis. Cancers 2019, 11, 1341. [CrossRef]23. Higgins, M.J.; Baselga, J. Targeted therapies for breast cancer. J. Clin. Investig. 2011, 121, 3797–3803. [CrossRef]24. Carrick, S.; Parker, S.; Thornton, C.E.; Ghersi, D.; Simes, J.; Wilcken, N. Single agent versus combination chemotherapy for metastatic breast cancer. Cochrane Database Syst. Rev. 2009, 2021, CD003372. [CrossRef]25. Osborne, C.K. Tamoxifen in the treatment of breast cancer. N. Engl. J. Med. 1998, 339, 1609–1618. [CrossRef] [PubMed]26. Smith, I.E.; Dowsett, M. Aromatase inhibitors in breast cancer. N. Engl. J. Med. 2003, 348, 2431–2442. [CrossRef] [PubMed]27. Spoerke, J.M.; Gendreau, S.; Walter, K.; Qiu, J.; Wilson, T.R.; Savage, H.; Aimi, J.; Derynck, M.K.; Chen, M.; Chan, I.T.; et al. Heterogeneity and clinical significance of ESR1 mutations in ER-positive metastatic breast cancer patients receiving fulvestrant. Nat. Commun. 2016, 7, 1–10. [CrossRef]28. Mayer, I.A.; Abramson, V.G.; Formisano, L.; Balko, J.M.; Estrada, M.V.; Sanders, M.E. A phase Ib study of alpelisib (BYL719), a PI3Kα-specific inhibitor, with letrozole in ER+/HER2− metastatic breast cancer. Clin. Cancer Res. 2017, 23, 26–34. [CrossRef]29. Coleman, R.L.; Brady, M.F.; Herzog, T.J.; Sabbatini, P.; Armstrong, D.K.; Walker, J.L.; Kim, B.G.; Fujiwara, K.; Tewari, K.S.; O’Malley, D.M.; et al. Bevacizumab and paclitaxel–carboplatin chemotherapy and secondary cytoreduction in recurrent, platinum-sensitiveovarian cancer (NRG Oncology/Gynecologic Oncology Group study GOG-0213): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2017, 18, 779–791. [CrossRef]30. Delaloge, S.; Pérol, D.; Courtinard, C.; Brain, E.; Asselain, B.; Bachelot, T.; Debled, M.; Dieras, V.; Campone, M.; Levy, C.; et al. Paclitaxel plus bevacizumab or paclitaxel as first-line treatment for HER2-negative metastatic breast cancer in a multicenter national observational study. Ann. Oncol. 2016, 27, 1725–1732. [CrossRef] [PubMed]31. Finn, R.S.; Martin, M.; Rugo, H.S.; Jones, S.; Im, S.-A.; Gelmon, K.; Harbeck, N.; Lipatov, O.N.; Walshe, J.M.; Moulder, S.; et al. Palbociclib and Letrozole in Advanced Breast Cancer. N. Engl. J. Med. 2016, 375, 1925–1936. [CrossRef]32. Turner, N.C.; Ro, J.; André, F.; Loi, S.; Verma, S.; Iwata, H.; Harbeck, N.; Loibl, S.; Bartlett, C.H.; Zhang, K.; et al. Palbociclib in Hormone-Receptor–Positive Advanced Breast Cancer. N. Engl. J. Med. 2015, 373, 209–219. [CrossRef]33. Hortobagyi, G.N.; Stemmer, S.M.; Burris, H.A.; Yap, Y.-S.; Sonke, G.S.; Paluch-Shimon, S.; Campone, M.; Blackwell, K.L.; André, F.; Winer, E.P.; et al. Ribociclib as First-Line Therapy for HR-Positive, Advanced Breast Cancer. N. Engl. J. Med. 2016, 375, 1738–1748.[CrossRef]34. Senkus, E.; Łacko, A. Over-treatment in metastatic breast cancer. Breast 2017, 31, 309–317. [CrossRef] [PubMed]35. Cardoso, F.; Harbeck, N.; Fallowfield, L.; Kyriakides, S.; Senkus, E. ESMO Guidelines Working Group. Locally recurrent or metastatic breast cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2012, 23, vii11–vii19. [CrossRef] [PubMed]36. Williams, M.; Lee, L.; Werfel, T.; Joly, M.M.M.; Hicks, D.J.; Rahman, B.; Elion, D.; McKernan, C.; Sanchez, V.; Estrada, M.V.; et al. Intrinsic apoptotic pathway activation increases response to anti-estrogens in luminal breast cancers. Cell Death Dis. 2018, 9, 1–14. [CrossRef] [PubMed]37. Richman, J.; Dowsett, M. Beyond 5 years: Enduring risk of recurrence in oestrogen receptor-positive breast cancer. Nat. Rev. Clin. Oncol. 2019, 16, 296–311. [CrossRef]38. Razavi, P.; Chang, M.T.; Xu, G.; Bandlamudi, C.; Ross, D.S.; Vasan, N.; Cai, Y.; Bielski, C.M.; Donoghue, M.T.A.; Jonsson, P.; et al. The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer Cell 2018, 34, 427–438.e6. [CrossRef]39. Boudreau, M.W.; Duraki, D.; Wang, L.; Mao, C.; Kim, J.E.; Henn, M.A.; Tang, B.; Fanning, S.W.; Kiefer, J.; Tarasow, T.M.; et al. A small-molecule activator of the unfolded protein response eradicates human breast tumors in mice. Sci. Transl. Med. 2021, 13, eabf1383. [CrossRef]40. Holland, S.J.; Pan, A.; Franci, C.; Hu, Y.; Chang, B.; Li, W.; Duan, M.; Torneros, A.; Yu, J.; Heckrodt, T.J.; et al. R428, a Selective Small Molecule Inhibitor of Axl Kinase, Blocks Tumor Spread and Prolongs Survival in Models of Metastatic Breast Cancer. Cancer Res. 2010, 70, 1544–1554. [CrossRef]41. Deng, S.; Krutilina, R.I.; Wang, Q.; Lin, Z.; Parke, D.N.; Playa, H.C.; Chen, H.; Miller, D.D.; Seagroves, T.N.; Li, W. An Orally Available Tubulin Inhibitor, VERU-111, Suppresses Triple-Negative Breast Cancer Tumor Growth and Metastasis and Bypasses Taxane Resistance. Mol. Cancer Ther. 2020, 19, 348–363. [CrossRef]42. Vernieri, C.; Corti, F.; Nichetti, F.; Ligorio, F.; Manglaviti, S.; Zattarin, E.; Rea, C.G.; Capri, G.; Bianchi, G.V.; De Braud, F. Everolimus versus alpelisib in advanced hormone receptor-positive HER2-negative breast cancer: Targeting different nodes of the PI3K/AKT/mTORC1 pathway with different clinical implications. Breast Cancer Res. 2020, 22, 1–13. [CrossRef]43. Jager, A.; De Vries, E.G.E.; Oordt, C.W.M.-V.D.H.V.; Neven, P.; Venema, C.M.; Glaudemans, A.W.J.M.; Wang, Y.; Bagley, R.G.; Conlan, M.G.; Aftimos, P. A phase 1b study evaluating the effect of elacestrant treatment on estrogen receptor availability and estradiol binding to the estrogen receptor in metastatic breast cancer lesions using 18F-FES PET/CT imaging. Breast Cancer Res. 2020, 22, 1–11. [CrossRef]44. Liang, J.; Zbieg, J.R.; Blake, R.A.; Chang, J.H.; Daly, S.; DiPasquale, A.G.; Friedman, L.S.; Gelzleichter, T.; Gill, M.; Giltnane, J.M.; et al. GDC-9545 (Giredestrant): A Potent and Orally Bioavailable Selective Estrogen Receptor Antagonist and Degrader with an Exceptional Preclinical Profile for ER+ Breast Cancer. J. Med. Chem. 2021, 64, 11841–11856. [CrossRef] [PubMed]45. Scott, J.S.; Moss, T.A.; Balazs, A.; Barlaam, B.; Breed, J.; Carbajo, R.J.; Chiarparin, E.; Davey, P.R.J.; Delpuech, O.; Fawell, S.; et al. Discovery of AZD9833, a Potent and Orally Bioavailable Selective Estrogen Receptor Degrader and Antagonist. J. Med. Chem. 2020, 63, 14530–14559. [CrossRef] [PubMed]46. Doll, R.; Peto, R. The Causes of Cancer: Quantitative Estimates of Avoidable Risks of Cancer in the United States Today. J. Natl. Cancer Inst. 1981, 66, 1192–1308. [CrossRef]47. Nicodemus, K.K.; Jacobsr, D.R., Jr.; Folsom, A.R. Whole and refined grain intake and risk of incident postmenopausal breast cancer (United States). Cancer Causes Control 2001, 12, 917–925. [CrossRef] [PubMed]48. Dydjow-Bendek, D.; Zagozdzon, P. Total Dietary Fats, Fatty Acids, and Omega-3/Omega-6 Ratio as Risk Factors of Breast Cancer in the Polish Population-a Case-Control Study. In Vivo 2020, 34, 423–431. [CrossRef]49. Tsilidis, K.K.; Travis, R.C.; Appleby, P.N.; Allen, N.E.; Lindström, S.; Albanes, D.; Ziegler, R.G.; McCullough, M.L.; Siddiq, A.; Barricarte, A.; et al. Insulin-like growth factor pathway genes and blood concentrations, dietary protein and risk of prostate cancer in the NCI Breast and Prostate Cancer Cohort Consortium (BPC3). Int. J. Cancer 2013, 133, 495–504. [CrossRef]50. Williams, C.M.; Dickerson, J.W. Dietary fat, hormones and breast cancer: The cell membrane as a possible site of interaction of these two risk factors. Eur. J. Surg. Oncol. 1987, 13, 89–104.51. Levi, F.; La Vecchia, C.; Gulie, C.; Negri, E. Dietary Factors and Breast-Cancer Risk in Vaud, Switzerland. Nutr. Cancer Int. J. 1993, 19, 327–335. [CrossRef]52. Mauvais-Jarvis, F.; Clegg, D.J.; Hevener, A.L. The Role of Estrogens in Control of Energy Balance and Glucose Homeostasis. Endocr. Rev. 2013, 34, 309–338. [CrossRef]53. Madak-Erdogan, Z.; Band, S.; Zhao, Y.C.; Smith, B.P.; Kulkoyluoglu-Cotul, E.; Zuo, Q.; Casiano, A.S.; Wrobel, K.; Rossi, G.; Smith, R.L.; et al. Free fatty acids rewire cancer metabolism in obesity-associated breast cancer via estrogen receptor and mTOR signaling. Cancer Res. 2019, 79, 2494–2510. [CrossRef]54. Zuo, Q.; Band, S.; Kesavadas, M.; Erdogan, Z.M. Obesity and Postmenopausal Hormone Receptor-positive Breast Cancer: Epidemiology and Mechanisms. Endocrinology 2021, 162, bqab195. [CrossRef] [PubMed]55. Jiralerspong, S.; Goodwin, P.J. Obesity and Breast Cancer Prognosis: Evidence, Challenges, and Opportunities. J. Clin. Oncol. 2016, 34, 4203–4216. [CrossRef] [PubMed]56. Suzuki, R.; Orsini, N.; Saji, S.; Key, T.J.; Wolk, A. Body weight and incidence of breast cancer defined by estrogen and progesterone receptor status-A meta-analysis. Int. J. Cancer 2009, 124, 698–712. [CrossRef] [PubMed]57. Iyengar, N.M.; Arthur, R.; Manson, J.E.; Chlebowski, R.T.; Kroenke, C.H.; Peterson, L.; Cheng, T.D.; Feliciano, E.; Lane, D.; Luo, J.; et al. Association of Body Fat and Risk of Breast Cancer in Postmenopausal Women with Normal Body Mass Index A Secondary Analysis of a Randomized Clinical Trial and Observational Study. JAMA Oncol. 2019, 5, 155–163. [CrossRef]58. Picon-Ruiz, M.; Morata-Tarifa, C.; Valle-Goffin, J.J.; Friedman, E.R.; Slingerland, J.M. Obesity and adverse breast cancer risk and outcome: Mechanistic insights and strategies for intervention. CA Cancer J. Clin. 2017, 67, 378–397. [CrossRef]59. Keum, N.; Greenwood, D.C.; Lee, D.H.; Kim, R.; Aune, D.; Ju, W.; Hu, F.B.; Giovannucci, E.L. Adult Weight Gain and Adiposity- Related Cancers: A Dose-Response Meta-Analysis of Prospective Observational Studies. J. Natl. Cancer Inst. 2015, 107, djv088.60. Renehan, A.G.; Tyson, M.; Egger, M.; Heller, R.F.; Zwahlen, M. Body-mass index and incidence of cancer: A systematic review and meta-analysis of prospective observational studies. Lancet 2008, 371, 569–578. [CrossRef]61. Chan, D.S.M.; Vieira, A.R.; Aune, D.; Bandera, E.V.; Greenwood, D.C.; McTiernan, A.; Rosenblatt, D.N.; Thune, I.; Vieira, R.; Norat, T. Body mass index and survival in women with breast cancer—systematic literature review and meta-analysis of 82 follow-up studies. Ann. Oncol. 2014, 25, 1901–1914. [CrossRef]62. Gathirua-Mwangi, W.G.; Palmer, J.R.; Champion, V.; Castro-Webb, N.; Stokes, A.C.; Adams-Campbell, L.; Marley, A.R.; Forman, M.R.; Rosenberg, L.; Bertrand, K.A. Maximum and Time-Dependent Body Mass Index and Breast Cancer Incidence Among Postmenopausal Women in the Black Women’s Health Study. Am. J. Epidemiol. 2022, 191, 646–654. [CrossRef]63. Chauhan, R.; Trivedi, V.; Rani, R.; Singh, U. A comparative analysis of body mass index with estrogen receptor, progesterone receptor and human epidermal growth factor receptor 2 status in pre- and postmenopausal breast cancer patients. J. Mid-Life Health 2020, 11, 210–216. [CrossRef]64. Statovci, D.; Aguilera, M.; Mac Sharry, J.; Melgar, S. The Impact of Western Diet and Nutrients on the Microbiota and Immune Response at Mucosal Interfaces. Front. Immunol. 2017, 8, 838. [CrossRef] [PubMed]65. Garcia-Montero, C.; Fraile-Martínez, O.; Gómez-Lahoz, A.M.; Pekarek, L.; Castellanos, A.J.; Noguerales-Fraguas, F.; Coca, S.; Guijarro, L.G.; García-Honduvilla, N.; Asúnsolo, A.; et al. Nutritional Components in Western Diet Versus Mediterranean Diet at the Gut Microbiota-Immune System Interplay. Implications for Health and Disease. Nutrients 2021, 13, 699. [CrossRef] [PubMed]66. Zinöcker, M.K.; Lindseth, I.A. The Western Diet–Microbiome-Host Interaction and Its Role in Metabolic Disease. Nutrients 2018, 10, 365. [CrossRef] [PubMed]67. Cully, M.; You, H.; Levine, A.J.; Mak, T.W. Beyond PTEN mutations: The PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nat. Rev. Cancer 2006, 6, 184–192. [CrossRef] [PubMed]68. Haythorne, E.; Rohm, M.; Van De Bunt, M.; Brereton, M.F.; Tarasov, A.I.; Blacker, T.S.; Sachse, G.; Dos Santos, M.S.; Exposito, R.T.; Davis, S.; et al. Diabetes causes marked inhibition of mitochondrial metabolism in pancreatic β-cells. Nat. Commun. 2019, 10, 1–17. [CrossRef]69. Newsholme, P.; Keane, K.N.; Carlessi, R.; Cruzat, V. Oxidative stress pathways in pancreatic β-cells and insulin-sensitive cells and tissues: Importance to cell metabolism, function, and dysfunction. Am. J. Physiol. Cell Physiol. 2019, 317, C420–C433. [CrossRef]70. Klement, R.J.; Kammerer, U. Is there a role for carbohydrate restriction in the treatment and prevention of cancer? Nutr. Metab. 2011, 8, 1–16. [CrossRef]71. LaPensee, C.R.; Hugo, E.R.; Ben-Jonathan, N. Insulin Stimulates Interleukin-6 Expression and Release in LS14 Human Adipocytes through Multiple Signaling Pathways. Endocrinology 2008, 149, 5415–5422. [CrossRef]72. Makino, T.; Noguchi, Y.; Yoshikawa, T.; Doi, C.; Nomura, K. Circulating interleukin 6 concentrations and insulin resistance in patients with cancer. Br. J. Surg. 1998, 85, 1658–1662. [CrossRef]73. Mccall, J.L.; Tuckey, J.A.; Parry, B.R. Serum Tumor-Necrosis-Factor-Alpha and Insulin Resistance in Gastrointestinal Cancer. Br. J. Surg. 1992, 79, 1361–1363. [CrossRef]74. Tian, M.; Zhang, H.; Nakasone, Y.; Mogi, K.; Endo, K. Expression of Glut-1 and Glut-3 in untreated oral squamous cell carcinoma compared with FDG accumulation in a PET study. Eur. J. Nucl. Med. Mol. Imaging 2004, 31, 5–12. [CrossRef] [PubMed]75. Zhang, H.-L.; Wang, M.-D.; Zhou, X.; Qin, C.-J.; Fu, G.-B.; Tang, L.; Wu, H.; Huang, S.; Zhao, L.-H.; Zeng, M.; et al. Blocking preferential glucose uptake sensitizes liver tumor-initiating cells to glucose restriction and sorafenib treatment. Cancer Lett. 2017, 388, 1–11. [CrossRef] [PubMed]76. Maldonado, R.; Talana, C.A.; Song, C.; Dixon, A.; Uehara, K.; Weichhaus, M. β-hydroxybutyrate does not alter the effects of glucose deprivation on breast cancer cells. Oncol. Lett. 2021, 21, 1. [CrossRef] [PubMed]77. Varghese, S.; Samuel, S.M.; Varghese, E.; Kubatka, P.; Büsselberg, D. High Glucose Represses the Anti-Proliferative and Pro- Apoptotic Effect of Metformin in Triple Negative Breast Cancer Cells. Biomolecules 2019, 9, 16. [CrossRef]78. Zuo, Q.; Mogol, A.N.; Liu, Y.-J.; Casiano, A.S.; Chien, C.; Drnevich, J.; Imir, O.B.; Kulkoyluoglu-Cotul, E.; Park, N.H.; Shapiro, D.J.; et al. Targeting metabolic adaptations in the breast cancer-liver metastatic niche using dietary approaches to improve endocrine therapy efficacy. Mol. Cancer Res. 2022, 20, 923–937. [CrossRef]79. Healy, M.E.; Chow, J.D.; Byrne, F.L.; Breen, D.S.; Leitinger, N.; Li, C.; Lackner, C.; Caldwell, S.H.; Hoehn, K.L. Dietary effects on liver tumor burden in mice treated with the hepatocellular carcinogen diethylnitrosamine. J. Hepatol. 2015, 62, 599–606. [CrossRef]80. Bechmann, L.P.; Hannivoort, R.A.; Gerken, G.; Hotamisligil, G.S.; Trauner, M.; Canbay, A. The interaction of hepatic lipid and glucose metabolism in liver diseases. J. Hepatol. 2012, 56, 952–964. [CrossRef]81. Zhong, X.-Y.; Yuan, X.-M.; Xu, Y.-Y.; Yin, M.; Yan, W.-W.; Zou, S.-W.; Wei, L.-M.; Lu, H.-J.; Wang, Y.-P.; Lei, Q.-Y. CARM1 Methylates GAPDH to Regulate Glucose Metabolism and Is Suppressed in Liver Cancer. Cell Rep. 2018, 24, 3207–3223. [CrossRef]82. Krstic, J.; Reinisch, I.; Schindlmaier, K.; Galhuber, M.; Riahi, Z.; Berger, N.; Kupper, N.; Moyschewitz, E.; Auer, M.; Michenthaler, H.; et al. Fasting improves therapeutic response in hepatocellular carcinoma through p53-dependent metabolic synergism. Sci. Adv. 2022, 8, eabh2635. [CrossRef]83. Wahdan-Alaswad, R.S.; Edgerton, S.M.; Salem, H.S.; Thor, A.D. Metformin Targets Glucose Metabolism in Triple Negative Breast Cancer. J. Oncol. Transl. Res. 2018, 4, 129. [CrossRef]84. Roy, R.; Hahm, E.R.; White, A.G.; Anderson, C.J.; Singh, S.V. AKT-dependent sugar addiction by benzyl isothiocyanate in breast cancer cells. Mol. Carcinog. 2019, 58, 996–1007. [CrossRef] [PubMed]85. Gluschnaider, U.; Hertz, R.; Ohayon, S.; Smeir, E.; Smets, M.; Pikarsky, E.; Bar-Tana, J. Long-Chain Fatty Acid Analogues Suppress Breast Tumorigenesis and Progression. Cancer Res. 2014, 74, 6991–7002. [CrossRef] [PubMed]86. Wei, M.; Brandhorst, S.; Shelehchi, M.; Mirzaei, H.; Cheng, C.W.; Budniak, J.; Groshen, S.; Mack, W.J.; Guen, E.; Di Biase, S.; et al. Fasting-mimicking diet and markers/risk factors for aging, diabetes, cancer, and cardiovascular disease. Sci. Transl. Med. 2017, 9. [CrossRef] [PubMed]87. Poff, A.; Ari, C.; Arnold, P.; Seyfried, T.; D’Agostino, D. Ketone supplementation decreases tumor cell viability and prolongs survival of mice with metastatic cancer. Int. J. Cancer 2014, 135, 1711–1720. [CrossRef] [PubMed]88. Allen, B.G.; Bhatia, S.K.; Anderson, C.M.; Eichenberger-Gilmore, J.M.; Sibenaller, Z.A.; Mapuskar, K.A.; Schoenfeld, J.D.; Buatti, J.M.; Spitz, D.R.; Fath, M.A. Ketogenic diets as an adjuvant cancer therapy: History and potential mechanism. Redox Biol. 2014, 2, 963–970. [CrossRef]89. Rigo, P.; Paulus, P.; Kaschten, B.J.; Hustinx, R.; Bury, T.; Jerusalem, G.; Benoit, T.; Foidart-Willems, J. Oncological applications ofpositron emission tomography with fluorine-18 fluorodeoxyglucose. Eur. J. Nucl. Med. 1996, 23, 1641–1674. [CrossRef]90. Kaji, K.; Nishimura, N.; Seki, K.; Sato, S.; Saikawa, S.; Nakanishi, K.; Furukawa, M.; Kawaratani, H.; Kitade, M.; Moriya, K.; et al. Sodium glucose cotransporter 2 inhibitor canagliflozin attenuates liver cancer cell growth and angiogenic activity by inhibiting glucose uptake. Int. J. Cancer 2018, 142, 1712–1722. [CrossRef]91. Zhang, X.; Qiao, Y.; Wu, Q.; Chen, Y.; Zou, S.; Liu, X.; Zhu, G.; Zhao, Y.; Chen, Y.; Yu, Y.; et al. The essential role of YAP O-GlcNAcylation in high-glucose-stimulated liver tumorigenesis. Nat. Commun. 2017, 8, 1–15. [CrossRef]92. Bu, P.; Chen, K.-Y.; Xiang, K.; Johnson, C.; Crown, S.B.; Rakhilin, N.; Ai, Y.; Wang, L.; Xi, R.; Astapova, I.; et al. Aldolase B-Mediated Fructose Metabolism Drives Metabolic Reprogramming of Colon Cancer Liver Metastasis. Cell Metab. 2018, 27, 1249–1262.e4. [CrossRef]93. Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [CrossRef]94. Anykin-Burns, N.; Ahmad, I.A.; Zhu, Y.; Oberley, L.W.; Spitz, D.R. Increased levels of superoxide and hydrogen peroxide mediate the differential susceptibility of cancer cells vs. Normal cells to glucose deprivation. Biochem. J. 2009, 418, 29–37. [CrossRef] [PubMed]95. Boros, L.G.; Lee, P.W.N.; Brandesa, J.L.; Cascante, M.; Muscarellaa, P.; Schirmera, W.J.; Melvina, W.S.; Ellisona, E.C. Nonoxidative pentose phosphate pathways and their direct role in ribose synthesis in tumors: Is cancer a disease of cellular glucose metabolism? Med. Hypotheses 1998, 50, 55–59.[CrossRef]96. Weber, D.D.; Aminazdeh-Gohari, S.; Kofler, B. Ketogenic diet in cancer therapy. Aging 2018, 10, 164–165. [CrossRef] [PubMed]97. Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [CrossRef] [PubMed]98. Buettner, G.R. Superoxide dismutase in redox biology: The roles of superoxide and hydrogen peroxide. Anti-Cancer Agents Med. Chem. 2011, 11, 341–346. [CrossRef] [PubMed]99. Veech, R.L. Ketone ester effects on metabolism and transcription. J. Lipid Res. 2014, 55, 2004–2006. [CrossRef]100. Hopkins, B.D.; Pauli, C.; Du, X.; Wang, D.G.; Li, X.; Wu, D.; Amadiume, S.C.; Goncalves, M.D.; Hodakoski, C.; Lundquist, M.R.; et al. Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature 2018, 560, 499–503, Erratum in Nature 2018, 563, E24. [CrossRef]101. Ho, V.W.; Leung, K.; Hsu, A.; Luk, B.; Lai, J.; Shen, S.Y.; Minchinton, A.I.; Waterhouse, D.; Bally, M.; Lin, W.; et al. A Low Carbohydrate, High Protein Diet Slows Tumor Growth and Prevents Cancer Initiation. Cancer Res. 2011, 71, 4484–4493. [CrossRef]102. Vanderheiden, M.G.; Cantley, L.C.; Thompson, C.B. UnderstandingtheWarburgEffect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [CrossRef]103. Byrne, F.L.;Hargett, S.R.; Lahiri, S.; Roy, R.J.; Berr, S. S.; Caldwell, S.H.; Hoehn, K.L. Serial MRI Imaging Reveals Minimal Impact of Ketogenic Diet on Established Liver Tumor Growth. Cancers 2018, 10, 312. [CrossRef]104. Tan-Shalaby, J. Ketogenic diets, and cancer: Emerging evidence.Fed.Pract.2017,34,37S.[PubMed]105. Allen, B.G.; Bhatia, S.K.; Buatti, J.; Brandt, K.E.; Lindholm, K.E.; Button, A.; Szweda, L.I.; Smith, B.J.; Spitz, D.R.; Fath, M.A. Ketogenic Diets Enhance Oxidative Stress and Radio-Chemo-Therapy Responses in Lung Cancer Xenografts. Clin. Cancer Res. 2013, 19, 3905–3913. [CrossRef] [PubMed]106.Abdelwahab, M.G.; Fenton, K.E.; Preul, M.C.; Rho, J.M.; Lynch,A.; Stafford, P.; Scheck, A.C. The Ketogenic Diet Isan Effective Adjuvant to Radiation Therapy for the Treatment of Malignant Glioma. PLoS ONE 2012, 7, e36197. [CrossRef] [PubMed]107. Fokidis, H.B.;Chin, M.Y.;Ho, V.W.;Adomat, H.H.; Soma,K.K.; Fazli,L.; Nip, K.M.; Cox, M.; Krystal, G.; Zoubeidi, A.; et. A low carbohydrate, high protein diet suppresses intratumoral androgen synthesis and slows castration-resistant prostate tumorgrowth in mice. J. Steroid Biochem. Mol. Biol. 2015, 150, 35–45. [CrossRef] [PubMed]108. Ho, V.W.; Hamilton, M.J.; Dang, N.-H.T.; Hsu, B.E.; Adomat, H.H.;Guns, E.S.; Weljie,A.; Samudio, I.; Bennewith, K.L.; Krystal, G. A low carbohydrate, high protein diet combined with celecoxib markedly reduces metastasis. Carcinogenesis 2014, 35, 2291–2299.[CrossRef] [PubMed]109. Martuscello,R.T.; Vedam-Mai,V.; McCarthy, D.J.; Schmoll, M.E.; Jundi, M.A.; Louviere, C.D.; Griffith, B.G.; Skinner, C.L.; Suslov, O.; Deleyrolle, L.P.; et al. A Supplemented High-Fat Low-Carbohydrate Diet for the Treatment of Glioblastoma. Clin. Cancer Res. 2016, 22, 2482–2495. [CrossRef]110. Caffa,I.; Spagnolo,V.; Vernieri,C.; Valdemarin,F.; Becherini,P.; Wei,M.; Brandhorst,S.; Zucal,C.; Driehuis,E.; Ferrando,L. Fasting-mimicking diet and hormone therapy induce breast cancer regression. Nature 2020, 583, 620–624. [CrossRef]111. Klement,R.J.;Champ,C.E.;Otto,C.;Kämmerer,U.Anti-TumorEffectsofKetogenicDietsinMice:AMeta-Analysis.PLoSONE 2016, 11, e0155050. [CrossRef]112. Yang, L.; TeSlaa, T.; Ng, S.; Nofal, M.; Wang, L.; Lan, T.; Zeng, X.; Cowan, A.; McBride, M.; Lu, W.; et al. Ketogenic diet and chemotherapy combine to disrupt pancreatic cancer metabolism and growth. Med 2022, 3, 119–136.e8. [CrossRef] [PubMed]113. DiBiase,S.; Shim,H.S.;Kim,K.H.; Vinciguerra,M.; Rappa,F.; Wei,M.; Brandhorst,S.; Cappello,F.; Mirzaei,H.; Lee,C.;etal. Fasting regulates EGR1 and protects from glucose-and dexamethasone-dependent sensitization to chemotherapy. PLoS Biol. 2017, 15, e2001951. [CrossRef]114. Salvadori,G.; Zanardi,F.; Iannelli,F.; Lobefaro,R.; Vernieri,C.;Longo,V.D.Fasting-mimicking diet blocks triple-negative breast cancer and cancer stem cell escape. Cell Metab. 2021, 33, 2247–2259.e6. [CrossRef] [PubMed]115. Elgendy,M.; Cirò,M.; Hosseini,A.; Weiszmann,J.; Mazzarella,L.;Ferrari,E.; Cazzoli,R.; Curigliano,G.; DeCensi,A.; Bonanni, B.; et al. Combination of Hypoglycemia and Metformin Impairs Tumor Metabolic Plasticity and Growth by Modulating the PP2A-GSK3β-MCL-1 Axis. Cancer Cell 2019, 35, 798–815.e5. [CrossRef] [PubMed]116. Puchalska,P.; Crawford,P.A. Multidimensional Rolesof Ketone Bodies in Fuel Metabolism, Signaling,and Therapeutics. Cell Metab. 2017, 25, 262–284. [CrossRef] [PubMed]117. Laffel,L. Ketone bodies: Are view of physiology, pathophysiology and application of monitoring to diabetes. Diabetes/Metab.Res. Rev. 1999, 15, 412–426. [CrossRef]118. Newman,J.C.; Verdin,E. β-Hydroxybutyrate: A Signaling Metabolite. Annu.Rev.Nutr.2017,37,51–76.[CrossRef]119. Dhillon, K.K.; Gupta, S.Biochemistry, Ketogenesis. InStat Pearls; Stat Pearls Publishing: TreasureIsland, FL, USA,2022.Available online: https://www.ncbi.nlm.nih.gov/books/NBK493179/ (accessed on 10 February 2022).120. Kim,D.Y.;Rho,J.M.Theketogenicdietandepilepsy.Curr.Opin.Clin.Nutr.Metab.Care2008,11,113–120.[CrossRef]121. Duan,Y.; Zhang,Y.; Dong,H.; Wang,Y.;Zhang,J. Effects of dietarypoly-beta-hydroxybutyrate(PHB) on microbiotacomposition and the mTOR signaling pathway in the intestines of litopenaeus vannamei. J. Microbiol. 2017, 55, 946–954. [CrossRef]122. Huang,C.; Wang,P.; Xu,X.; Zhang,Y.; Gong,Y.; Hu,W.; Gao,M.; Wu,Y.; Ling,Y.; Zhao,X.; etal.Theketonebodymetabolite beta-hydroxybutyrate induces an antidepression-associated ramification of microglia via HDACs inhibition-triggered Akt-small RhoGTPase activation. Glia 2018, 66, 256–278. [CrossRef]123. Vernieri, C.; Fucà, G.; Ligorio, F.; Huber, V.; Vingiani, A.; Iannelli, F.; Raimondi, A.; Rinchai, D.; Frigè, G.; Belfiore, A.; et al. Fasting-Mimicking Diet Is Safe and Reshapes Metabolism and Antitumor Immunity in Patients with Cancer. Cancer Discov. 2022, 12, 90–107. [CrossRef]124. Han, Y.-M.; Ramprasath, T.; Zou, M.-H. β-hydroxybutyrate and its metabolic effects on age-associated pathology.Exp.Mol.Med. 2020, 52, 548–555. [CrossRef]125. Kim, D.H.; Park, M.H.; Ha, S.; Bang, E.J.; Lee, Y.; Lee, A.K.; Lee, J.; Yu, B.P.; Chung, H.Y. Anti-inflammatory action of beta-hydroxybutyrate via modulation of PGC-1alpha and FoxO1, mimicking calorie restriction. Aging 2019, 11, 1283–1304. [CrossRef]126. Shimazu,T.; Hirschey,M.D.; Newman,J.; He,W.; Shirakawa,K.; LeMoan,N.; Grueter,C.A.; Lim,H.; Saunders,L.R.; Stevens, R.D.; et al. Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 2013, 339, 211–214. [CrossRef] [PubMed]127. Pant,K.; Peixoto,E.; Richard,S.; Gradilone,S.A. Role of Histone Deacetylases in Carcinogenesis: Potential Role in Cholangiocarcinoma. Cells 2020, 9, 780. [CrossRef]128. Martinez-Outschoorn, U.E.; Prisco, M.; Ertel, A.; Tsirigos, A.; Lin, Z.; Pavlides, S.; Wang, C.; Flomenberg, N.; Knudsen, E.S.; Howell, A. Ketones and lactate increase cancer cell “stemness,” driving recurrence, metastasis and poor clinical outcome in breast cancer: Achieving personalized medicine via Metabolo-Genomics. Cell Cycle 2011, 10, 1271–1286. [CrossRef] [PubMed]129. Bonuccelli,G.; Tsirigos,A.; Whitaker-Menezes,D.; Pavlides,S.; Pestell,R.G.; Chiavarina,B.; Frank,P.G.; Flomenberg,N.; Howell, A.; Martinez-Outschoorn, U.E.; et al. Ketones and lactate “fuel” tumor growth and metastasis: Evidence that epithelial cancer cells use oxidative mitochondrial metabolism. Cell Cycle 2010, 9, 3506–3514. [CrossRef] [PubMed]130. Huang,C.; Chang,P.; Kuo,W.; Chen,C.; Jeng,Y.; Chang,K.; Shew,J.; Hu,C.; Lee,W. Adipocytes promote malignant growth of breast tumours with monocarboxylate transporter 2 expression via beta-hydroxybutyrate. Nat. Commun. 2017, 8, 14706. [CrossRef] [PubMed]131. Rodrigues, L.M.; Uribe-Lewis, S.; Madhu, B.; Honess, D.J.; Stubbs, M.; Griffiths, J.R. The action of β-hydroxybutyrate on the growth, metabolism and global histone H3 acetylation of spontaneous mouse mammary tumours: Evidence of a β- hydroxybutyrate paradox. Cancer Metab. 2017, 5, 1–13. [CrossRef]132. Shukla,S.K.; Gebregiworgis,T.; Purohit,V.; Chaika,N.V.; Gunda,V.;Radhakrishnan,P.; Mehla,K.; Pipinos,I.I.;Powers,R.; Yu, F.; et al. Metabolic reprogramming induced by ketone bodies diminishes pancreatic cancer cachexia. Cancer Metab. 2014, 2, 18. [CrossRef]133. Mikami, D.; Kobayashi, M.; Uwada, J.; Yazawa, T.; Kamiyama, K.; Nishimori, K.; Nishikawa, Y.; Nishikawa, S.; Yokoi, S.; Taniguchi, T.; et al. β-Hydroxybutyrate enhances the cytotoxic effect of cisplatin via the inhibition of HDAC/survivin axis in human hepatocellular carcinoma cells. J. Pharmacol. Sci. 2020, 142, 1–8. [CrossRef]134. Schmidt,M.; Pfetzer,N.; Schwab,M.; Strauss,I.; Kämmerer,U. Effects of a ketogenic diet on the quality of life in 16 patients with advanced cancer: A pilot trial. Nutr. Metab. 2011, 8, 54. [CrossRef]135. Bartmann,C.; Raman,S.R.J.; Flöter,J.; Schulze,A.; Bahlke,K.; Willingstorfer,J.; Strunz,M.; Wöckel,A.; Klement,R.J.; Kapp, M.; et al. Beta-hydroxybutyrate (3-OHB) can influence the energetic phenotype of breast cancer cells, but does not impact their proliferation and the response to chemotherapy or radiation. Cancer Metab. 2018, 6, 8. [CrossRef] [PubMed]136. Donohoe,D.R.; Collins,L.B.; Wali,A.; Bigler,R.; Sun,W.; Bultman,S.J. The Warburg Effect Dictates the Mechanism of Butyrate-Mediated Histone Acetylation and Cell Proliferation. Mol. Cell 2012, 48, 612–626. [CrossRef] [PubMed]137. Lupton,J.R. Microbial Degradation Products Influence Colon Cancer Risk: The Butyrate Controversy.J.Nutr.2004,134,479–482. [CrossRef]138. Luo,S.; Li,Z.; Mao,L.; Chen,S.; Sun,S. Sodium butyrate induces autophagy incolorectal cancer cells through LKB1/AMPK signaling. J. Physiol. Biochem. 2019, 75, 53–63. [CrossRef] [PubMed]139. Salimi,V.; Shahsavari,Z.; Safizadeh,B.; Hosseini,A.;Khademian,N.;Tavakoli-Yaraki,M. Sodium butyrate promotes apoptosis in breast cancer cells through reactive oxygen species (ROS) formation and mitochondrial impairment. Lipids Health Dis. 2017, 16, 1–11. [CrossRef] [PubMed]140. Vernieri, C.; Ligorio, F.; Zattarin, E.; Rivoltini, L.; De Braud, F. Fasting-mimicking diet plus chemotherapy in breast cancer treatment. Nat. Commun. 2020, 11, 1–4. [CrossRef]141. DeGroot,S.; Lugtenberg,R.T.; Cohen,D.; Welters,M.J.P.; Ehsan,I.; Vreeswijk,M.P.G.; Smit,V.T.H.B.M.; deGraaf,H.; Heijns,J.B.; Portielje, J.E.A.; et al. Fasting mimicking diet as an adjunct to neoadjuvant chemotherapy for breast cancer in the multicentre randomized phase 2 DIRECT trial. Nat. Commun. 2020, 11, 1–9. [CrossRef]142. DiBiase,S.; Lee,C.; Brandhorst,S.; Manes,B.; Buono,R.; Cheng,C.-W.; Cacciottolo,M.; Martin-Montalvo,A.; DeCabo,R.; Wei, M.; et al. Fasting-Mimicking Diet Reduces HO-1 to Promote T Cell-Mediated Tumor Cytotoxicity. Cancer Cell 2016, 30, 136–146. [CrossRef]Tarver, T. Breast Cancer Facts & Figures 2021; American Cancer Society: Atlanta, GA, USA, 2021.
- National Cancer Institute. Cancer Stat Facts: Female Breast Cancer; National Cancer Institute: Bethesda, MD, USA, 2020.
- Nasrindokht Azamjah; Yasaman Soltan-Zadeh; Farid Zayeri; Global Trend of Breast Cancer Mortality Rate: A 25-Year Study. Asian Pacific Journal of Cancer Prevention 2019, 20, 2015-2020, 10.31557/APJCP.2019.20.7.2015.
- Sarah Lévesque; Jonathan G. Pol; Gladys Ferrere; Lorenzo Galluzzi; Laurence Zitvogel; Guido Kroemer; Trial watch: dietary interventions for cancer therapy. OncoImmunology 2019, 8, e1591878, 10.1080/2162402x.2019.1591878.
- Angela B. Mariotto; Ruth Etzioni; Marc Hurlbert; Lynne Penberthy; Musa Mayer; Estimation of the Number of Women Living with Metastatic Breast Cancer in the United States. Cancer Epidemiology, Biomarkers & Prevention 2017, 26, 809-815, 10.1158/1055-9965.epi-16-0889.
- Gaorav P. Gupta; Joan Massagué; Cancer Metastasis: Building a Framework. Cell 2006, 127, 679-695, 10.1016/j.cell.2006.11.001.
- Rebecca L. Siegel; Kimberly D. Miller; Hannah E. Fuchs; Ahmedin Jemal; Cancer Statistics, 2021. CA: A Cancer Journal for Clinicians 2021, 71, 7-33, 10.3322/caac.21654.
- Lee Guek Eng; Shaheenah Dawood; Victoria Sopik; Benjamin Haaland; Pui San Tan; Nirmala Bhoo-Pathy; Ellen Warner; Javaid Iqbal; Steven A. Narod; Rebecca Dent; et al. Ten-year survival in women with primary stage IV breast cancer. Breast Cancer Research and Treatment 2016, 160, 145-152, 10.1007/s10549-016-3974-x.
- SEER Cancer Statistics Review, 1975–2017 . National Cancer Institute. Retrieved 2022-7-20
- Do Abha Soni; Zhiyong Ren; Omar Hameed; Diptiman Chanda; Charity J Morgan; Gene P. Siegal; Shi Wei; Breast Cancer Subtypes Predispose the Site of Distant Metastases. American Journal of Clinical Pathology 2015, 143, 471-478, 10.1309/ajcpyo5fsv3upexs.
- Yue Gong; Yi-Rong Liu; Peng Ji; Xin Hu; Zhi-Ming Shao; Impact of molecular subtypes on metastatic breast cancer patients: a SEER population-based study. Scientific Reports 2017, 7, srep45411, 10.1038/srep45411.
- Samantha R Horn; Kelsey C Stoltzfus; Eric J Lehrer; Laura A. Dawson; Leila Tchelebi; Niraj J Gusani; Navesh K Sharma; Hanbo Chen; Daniel M Trifiletti; Nicholas G Zaorsky; et al. Epidemiology of liver metastases. Cancer Epidemiology 2020, 67, 101760, 10.1016/j.canep.2020.101760.
- Narmeen S. Rashid; Jacqueline M. Grible; Charles V. Clevenger; J. Chuck Harrell; Breast cancer liver metastasis: current and future treatment approaches. Clinical & Experimental Metastasis 2021, 38, 263-277, 10.1007/s10585-021-10080-4.
- René Adam; Thomas Aloia; Jinane Krissat; Marie-Pierre Bralet; Bernard Paule; Sylvie Giacchetti; Valerie Delvart; Daniel Azoulay; Henri Bismuth; Denis Castaing; et al. Is Liver Resection Justified for Patients With Hepatic Metastases From Breast Cancer?. Annals of Surgery 2006, 244, 897-908, 10.1097/01.sla.0000246847.02058.1b.
- Jannemarie de Ridder; Johannes H. W. de Wilt; Femke Simmer; Lucy Overbeek; Valery Lemmens; Iris Nagtegaal; Incidence and origin of histologically confirmed liver metastases: an explorative case-study of 23,154 patients. Oncotarget 2016, 7, 55368-55376, 10.18632/oncotarget.10552.
- Margaret C Cummings; Peter T Simpson; Lynne E Reid; Janani Jayanthan; Joanna Skerman; Sarah Song; Amy E McCart Reed; Jamie R Kutasovic; Adrienne L Morey; Louise Marquart; et al.Peter O'RourkeSunil R Lakhani Metastatic progression of breast cancer: insights from 50 years of autopsies. The Journal of Pathology 2013, 232, 23-31, 10.1002/path.4288.
- Lei Ji; Lei Cheng; Xiuzhi Zhu; Yu Gao; Lei Fan; Zhonghua Wang; Risk and prognostic factors of breast cancer with liver metastases. BMC Cancer 2021, 21, 1-15, 10.1186/s12885-021-07968-5.
- Jingjing Xie; Zhongyuan Xu; A Population-Based Study on Liver Metastases in Women with Newly Diagnosed Breast Cancer. Cancer Epidemiology, Biomarkers & Prevention 2019, 28, 283-292, 10.1158/1055-9965.epi-18-0591.
- Jennifer R Diamond; Christina A Finlayson; Virginia F Borges; Hepatic complications of breast cancer. The Lancet Oncology 2009, 10, 615-621, 10.1016/s1470-2045(09)70029-4.
- V Patanaphan; O M Salazar; R Risco; Breast cancer: metastatic patterns and their prognosis.. Southern Medical Journal 1988, 81, 1109–1112.
- Rui Cao; Li-Ping Wang; Serological Diagnosis of Liver Metastasis in Patients with Breast Cancer. Cancer Biology & Medicine 2012, 9, 57-62, 10.3969/j.issn.2095-3941.2012.01.011.
- Reto Bale; Daniel Putzer; Peter Schullian; Local Treatment of Breast Cancer Liver Metastasis. Cancers 2019, 11, 1341, 10.3390/cancers11091341.
- Michaela J. Higgins; José Baselga; Targeted therapies for breast cancer. Journal of Clinical Investigation 2011, 121, 3797-3803, 10.1172/jci57152.
- Sue Carrick; Sharon Parker; Charlene E Thornton; Davina Ghersi; John Simes; Nicholas Wilcken; Single agent versus combination chemotherapy for metastatic breast cancer. Cochrane Database of Systematic Reviews 2009, 2021, CD003372, 10.1002/14651858.cd003372.pub3.
- C. Kent Osborne; Tamoxifen in the Treatment of Breast Cancer. New England Journal of Medicine 1998, 339, 1609-1618, 10.1056/nejm199811263392207.
- Ian E. Smith; Mitch Dowsett; Aromatase Inhibitors in Breast Cancer. New England Journal of Medicine 2003, 348, 2431-2442, 10.1056/nejmra023246.
- Jill M. Spoerke; Steven Gendreau; Kimberly Walter; Jiaheng Qiu; Timothy R. Wilson; Heidi Savage; Junko Aimi; Mika K. Derynck; Meng Chen; Iris T. Chan; et al.Lukas C. AmlerGarret M. HamptonStephen JohnstonIan KropPeter SchmidMark R. Lackner Heterogeneity and clinical significance of ESR1 mutations in ER-positive metastatic breast cancer patients receiving fulvestrant. Nature Communications 2016, 7, 11579, 10.1038/ncomms11579.
- Ingrid A. Mayer; Vandana G. Abramson; Luigi Formisano; Justin M. Balko; Mónica V. Estrada; Melinda E. Sanders; Dejan Juric; David Solit; Michael F. Berger; Helen H. Won; et al.Yisheng LiLewis C. CantleyEric WinerCarlos L. Arteaga A Phase Ib Study of Alpelisib (BYL719), a PI3Kα-Specific Inhibitor, with Letrozole in ER+/HER2− Metastatic Breast Cancer. Clinical Cancer Research 2016, 23, 26-34, 10.1158/1078-0432.ccr-16-0134.
- Robert L Coleman; Mark F Brady; Thomas J Herzog; Paul Sabbatini; Deborah K Armstrong; Joan L Walker; Byoung-Gie Kim; Keiichi Fujiwara; Krishnansu S Tewari; David M O'Malley; et al.Susan A DavidsonStephen C RubinPaul DiSilvestroKaren Basen-EngquistHelen HuangJohn K ChanNick M SpirtosRaheela AshfaqRobert S Mannel Bevacizumab and paclitaxel–carboplatin chemotherapy and secondary cytoreduction in recurrent, platinum-sensitive ovarian cancer (NRG Oncology/Gynecologic Oncology Group study GOG-0213): a multicentre, open-label, randomised, phase 3 trial. The Lancet Oncology 2017, 18, 779-791, 10.1016/s1470-2045(17)30279-6.
- S. Delaloge; D. Pérol; C. Courtinard; E. Brain; B. Asselain; T. Bachelot; M. Debled; V. Dieras; M. Campone; C. Levy; et al.W. JacotV. LorgisC. VeyretF. DalencJ. M. FerreroL. UwerP. KerbratA. GoncalvesM. A. Mouret-ReynierT. PetitC. JouannaudL. VanlemmensG. ChenucT. GuesmiaM. RobainC. Cailliot Paclitaxel plus bevacizumab or paclitaxel as first-line treatment for HER2-negative metastatic breast cancer in a multicenter national observational study. Annals of Oncology 2016, 27, 1725-1732, 10.1093/annonc/mdw260.
- Richard S. Finn; Miguel Martin; Hope S. Rugo; Stephen Jones; Seock-Ah Im; Karen Gelmon; Nadia Harbeck; Oleg N. Lipatov; Janice M. Walshe; Stacy Moulder; et al.Eric GauthierDongrui R. LuSophia RandolphVéronique DiérasDennis J. Slamon Palbociclib and Letrozole in Advanced Breast Cancer. New England Journal of Medicine 2016, 375, 1925-1936, 10.1056/nejmoa1607303.
- Nicholas C. Turner; Jungsil Ro; Fabrice André; Sherene Loi; Sunil Verma; Hiroji Iwata; Nadia Harbeck; Sibylle Loibl; Cynthia Huang Bartlett; Ke Zhang; et al.Carla GiorgettiSophia RandolphMaria KoehlerMassimo Cristofanilli Palbociclib in Hormone-Receptor–Positive Advanced Breast Cancer. New England Journal of Medicine 2015, 373, 209-219, 10.1056/nejmoa1505270.
- Gabriel N. Hortobagyi; Salomon M. Stemmer; Howard A. Burris; Yoon-Sim Yap; Gabe S. Sonke; Shani Paluch-Shimon; Mario Campone; Kimberly L. Blackwell; Fabrice André; Eric P. Winer; et al.Wolfgang JanniSunil VermaPierfranco ConteCarlos L. ArteagaDavid A. CameronKatarina PetrakovaLowell L. HartCristian VillanuevaArlene ChanErik JakobsenArnd NuschOlga BurdaevaEva-Maria GrischkeEmilio AlbaErik WistNorbert MarschnerAnne M. FavretDenise YardleyThomas BachelotLing-Ming TsengSibel BlauFengjuan XuanFarida SouamiMichelle MillerCaroline GermaSamit HirawatJoyce O’Shaughnessy Ribociclib as First-Line Therapy for HR-Positive, Advanced Breast Cancer. New England Journal of Medicine 2016, 375, 1738-1748, 10.1056/nejmoa1609709.
- Elżbieta Senkus; Aleksandra Łacko; Over-treatment in metastatic breast cancer. The Breast 2017, 31, 309-317, 10.1016/j.breast.2016.06.024.
- Fatima Cardoso; N. Harbeck; L. Fallowfield; S. Kyriakides; Elżbieta Senkus; Locally recurrent or metastatic breast cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Annals of Oncology 2012, 23, vii11-vii19, 10.1093/annonc/mds232.
- Michelle Williams; Linus Lee; Thomas Werfel; Meghan M. Morrison Joly; Donna J. Hicks; Bushra Rahman; David Elion; Courtney McKernan; Violeta Sanchez; Monica Valeria Estrada; et al.Suleiman MassarwehRichard ElledgeCraig DuvallRebecca S. Cook Intrinsic apoptotic pathway activation increases response to anti-estrogens in luminal breast cancers. Cell Death & Disease 2018, 9, 1-14, 10.1038/s41419-017-0072-x.
- Juliet Richman; Mitch Dowsett; Beyond 5 years: enduring risk of recurrence in oestrogen receptor-positive breast cancer. Nature Reviews Clinical Oncology 2018, 16, 296-311, 10.1038/s41571-018-0145-5.
- Pedram Razavi; Matthew T. Chang; Guotai Xu; Chaitanya Bandlamudi; Dara S. Ross; Neil Vasan; Yanyan Cai; Craig M. Bielski; Mark T.A. Donoghue; Philip Jonsson; et al.Alexander PensonRonglai ShenFresia ParejaRitika KundraSumit MiddhaMichael L. ChengAhmet ZehirCyriac KandothRuchi PatelKety HubermanLillian M. SmythKomal JhaveriShanu ModiTiffany A. TrainaChau DangWen ZhangBritta WeigeltBob T. LiMarc LadanyiDavid M. HymanNikolaus SchultzMark E. RobsonClifford HudisEdi BrogiAgnes VialeLarry NortonMaura N. DicklerMichael F. BergerChristine A. Iacobuzio-DonahueSarat ChandarlapatyMaurizio ScaltritiJorge S. Reis-FilhoDavid B. SolitBarry S. TaylorJosé Baselga The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer Cell 2018, 34, 427-438.e6, 10.1016/j.ccell.2018.08.008.
- Matthew W. Boudreau; Darjan Duraki; Lawrence Wang; Chengjian Mao; Ji Eun Kim; Madeline A. Henn; Bingtao Tang; Sean W. Fanning; Jeffrey Kiefer; Theodore M. Tarasow; et al.Elizabeth M. BruckheimerRamon MorenoSpyro MoussesGeoffrey L. GreeneEdward J. RoyBen Ho ParkTimothy M. FanErik R. NelsonPaul J. HergenrotherDavid J. Shapiro A small-molecule activator of the unfolded protein response eradicates human breast tumors in mice. Science Translational Medicine 2021, 13, 1383, 10.1126/scitranslmed.abf1383.
- Sacha J. Holland; Alison Pan; Christian Franci; Yuanming Hu; Betty Chang; Weiqun Li; Matt Duan; Allan Torneros; Jiaxin Yu; Thilo J. Heckrodt; et al.Jing ZhangPingyu DingAyodele ApatiraJoanne ChuaRalf BrandtPolly PineDane GoffRajinder SinghDonald G. PayanYasumichi Hitoshi R428, a Selective Small Molecule Inhibitor of Axl Kinase, Blocks Tumor Spread and Prolongs Survival in Models of Metastatic Breast Cancer. Cancer Research 2010, 70, 1544-1554, 10.1158/0008-5472.can-09-2997.
- Shanshan Deng; Raisa I. Krutilina; Qinghui Wang; Zongtao Lin; Deanna N. Parke; Hilaire C. Playa; Hao Chen; Duane D. Miller; Tiffany N. Seagroves; Wei Li; et al. An Orally Available Tubulin Inhibitor, VERU-111, Suppresses Triple-Negative Breast Cancer Tumor Growth and Metastasis and Bypasses Taxane Resistance. Molecular Cancer Therapeutics 2020, 19, 348-363, 10.1158/1535-7163.mct-19-0536.
- Claudio Vernieri; Francesca Corti; Federico Nichetti; Francesca Ligorio; Sara Manglaviti; Emma Zattarin; Carmen G. Rea; Giuseppe Capri; Giulia V. Bianchi; Filippo De Braud; et al. Everolimus versus alpelisib in advanced hormone receptor-positive HER2-negative breast cancer: targeting different nodes of the PI3K/AKT/mTORC1 pathway with different clinical implications. Breast Cancer Research 2020, 22, 1-13, 10.1186/s13058-020-01271-0.
- Agnes Jager; Elisabeth G. E. De Vries; C. Willemien Menke-Van Der Houven Van Oordt; Patrick Neven; Clasina M. Venema; Andor W. J. M. Glaudemans; Yamei Wang; Rebecca G. Bagley; Maureen G. Conlan; Philippe Aftimos; et al. A phase 1b study evaluating the effect of elacestrant treatment on estrogen receptor availability and estradiol binding to the estrogen receptor in metastatic breast cancer lesions using 18F-FES PET/CT imaging. Breast Cancer Research 2020, 22, 1-11, 10.1186/s13058-020-01333-3.
- Jun Liang; Jason R. Zbieg; Robert A. Blake; Jae H. Chang; Stephen Daly; Antonio G. DiPasquale; Lori S. Friedman; Thomas Gelzleichter; Matthew Gill; Jennifer M. Giltnane; et al.Simon GoodacreJane GuanSteven J. HartmanEllen Rei IngallaLorn KategayaJames R. KieferTracy KleinheinzSharada S. LabadieTommy LaiJun LiJiangpeng LiaoZhiguo LiuVidhi ModyNeville McLeanCiara MetcalfeMichelle A. NanniniJason OehMartin G. O’RourkeDaniel F. OrtwineYingqing RanNicholas C. RayFabien RousselAmy SambroneDeepak SampathLeah K. SchuttMaia VinogradovaJohn WaiTao WangIngrid E. WertzJonathan R. WhiteSiew Kuen YeapAmy YoungBirong ZhangXiaoping ZhengWei ZhouYu ZhongXiaojing Wang GDC-9545 (Giredestrant): A Potent and Orally Bioavailable Selective Estrogen Receptor Antagonist and Degrader with an Exceptional Preclinical Profile for ER+ Breast Cancer. Journal of Medicinal Chemistry 2021, 64, 11841-11856, 10.1021/acs.jmedchem.1c00847.
- James S. Scott; Thomas A. Moss; Amber Balazs; Bernard Barlaam; Jason Breed; Rodrigo J. Carbajo; Elisabetta Chiarparin; Paul R. J. Davey; Oona Delpuech; Stephen Fawell; et al.David I. FisherSladjana GagricaEric T. GanglTyler GrebeRyan David Robert GreenwoodSudhir HandeHolia Hatoum-MokdadKara HerlihySamantha HughesThomas A. HuntHoan HuynhSophie L. M. JanbonTony JohnsonStefan KavanaghTeresa C. M. KlinowskaMandy LawsonAndrew S. ListerStacey MardenDermot F. McGinnityChristopher J. MorrowJ. Willem M. NissinkDaniel H. O’DonovanBo PengRadoslaw PolanskiDarren S. SteadStephen StokesKumar ThakurScott R. ThronerMichael J. TuckerJeffrey G. VarnesHaixia WangDavid M. WilsonDedong WuYe WuBin YangWenzhan Yang Discovery of AZD9833, a Potent and Orally Bioavailable Selective Estrogen Receptor Degrader and Antagonist. Journal of Medicinal Chemistry 2020, 63, 14530-14559, 10.1021/acs.jmedchem.0c01163.
- Richard Doll; Richard Peto; The Causes of Cancer: Quantitative Estimates of Avoidable Risks of Cancer in the United States Today. JNCI: Journal of the National Cancer Institute 1981, 66, 1192-1308, 10.1093/jnci/66.6.1192.
- Kristin K. Nicodemus; David R. Jacobs Jr; Aaron R. Folsom; Whole and refined grain intake and risk of incident postmenopausal breast cancer (United States). Cancer Causes & Control 2000, 12, 917-925, 10.1023/a:1013746719385.
- Dorota Dydjow-Bendek; Pawel Zagoźdźon; Total Dietary Fats, Fatty Acids, and Omega-3/Omega-6 Ratio as Risk Factors of Breast Cancer in the Polish Population – a Case-Control Study. In Vivo 2019, 34, 423-431, 10.21873/invivo.11791.
- Konstantinos K. Tsilidis; Ruth C. Travis; Paul N. Appleby; Naomi E. Allen; Sara Lindström; Demetrius Albanes; Regina G. Ziegler; Marjorie L. McCullough; Afshan Siddiq; Aurelio Barricarte; et al.Sonja I. BerndtH. Bas Bueno-De-MesquitaStephen J. ChanockE. David CrawfordW. Ryan DiverSusan M. GapsturEdward GiovannucciFangyi GuChristopher A. HaimanRichard B. HayesDavid J. HunterMattias JohanssonRudolf KaaksLaurence N. KolonelPeter KraftLoic Le MarchandKim OvervadSilvia PolidoroElio RiboliFredrick R. SchumacherVictoria L. StevensDimitrios TrichopoulosJarmo VirtamoWalter C. WillettTimothy J Key Insulin-like growth factor pathway genes and blood concentrations, dietary protein and risk of prostate cancer in the NCI Breast and Prostate Cancer Cohort Consortium (BPC3). International Journal of Cancer 2013, 133, 495-504, 10.1002/ijc.28042.
- C M Williams; J W Dickerson; Dietary fat, hormones and breast cancer: the cell membrane as a possible site of interaction of these two risk factors.. European Journal of Surgical Oncology 1987, 13, 89-104.
- Fabio Levi; Carlo La Vecchia; Cristina Gulie; Eva Negri; Dietary factors and breast cancer risk in Vaud, Switzerland. Nutrition and Cancer 1992, 19, 327-335, 10.1080/01635589309514263.
- Franck Mauvais-Jarvis; Deborah J. Clegg; Andrea L. Hevener; The Role of Estrogens in Control of Energy Balance and Glucose Homeostasis. Endocrine Reviews 2013, 34, 309-338, 10.1210/er.2012-1055.
- Zeynep Madak-Erdogan; Shoham Band; Yiru Chen Zhao; Brandi Patrice Smith; Eylem Kulkoyluoglu-Cotul; Qianying Zuo; Ashlie Santaliz Casiano; Kinga Wrobel; Gianluigi Rossi; Rebecca Lee Smith; et al.Sung Hoon KimJohn A. KatzenellenbogenMariah L. JohnsonMeera PatelNatascia MarinoAnna Maria V. StornioloJodi A. Flaws Free Fatty Acids Rewire Cancer Metabolism in Obesity-Associated Breast Cancer via Estrogen Receptor and mTOR Signaling. Cancer Research 2019, 79, 2494-2510, 10.1158/0008-5472.can-18-2849.
- Qianying Zuo; Shoham Band; Mrinali Kesavadas; Zeynep Madak Erdogan; Obesity and Postmenopausal Hormone Receptor-positive Breast Cancer: Epidemiology and Mechanisms. Endocrinology 2021, 162, 195, 10.1210/endocr/bqab195.
- Sao Jiralerspong; Pamela Goodwin; Obesity and Breast Cancer Prognosis: Evidence, Challenges, and Opportunities. Journal of Clinical Oncology 2016, 34, 4203-4216, 10.1200/jco.2016.68.4480.
- Reiko Suzuki; Nicola Orsini; Shigehira Saji; Timothy J. Key; Alicja Wolk; Body weight and incidence of breast cancer defined by estrogen and progesterone receptor status-A meta-analysis. International Journal of Cancer 2009, 124, 698-712, 10.1002/ijc.23943.
- Neil M. Iyengar; Rhonda Arthur; JoAnn E. Manson; Rowan T. Chlebowski; Candyce H. Kroenke; Lindsay Peterson; Ting-Yuan D. Cheng; Elizabeth C. Feliciano; Dorothy Lane; Juhua Luo; et al.Rami NassirKathy PanSylvia Wassertheil-SmollerVictor KamenskyThomas E. RohanAndrew J. Dannenberg Association of Body Fat and Risk of Breast Cancer in Postmenopausal Women With Normal Body Mass Index. JAMA Oncology 2019, 5, 155-163, 10.1001/jamaoncol.2018.5327.
- Manuel Picón Ruiz; Cynthia Morata Tarifa; Janeiro J. Valle-Goffin; Eitan R. Friedman; Joyce M. Slingerland; Obesity and adverse breast cancer risk and outcome: Mechanistic insights and strategies for intervention. CA: A Cancer Journal for Clinicians 2017, 67, 378-397, 10.3322/caac.21405.
- Nana Keum; Darren C Greenwood; Dong Hoon Lee; Rockli Kim; Dagfinn Aune; Woong Ju; Frank B Hu; Edward L Giovannucci; Adult Weight Gain and Adiposity-Related Cancers: A Dose-Response Meta-Analysis of Prospective Observational Studies. Journal of the National Cancer Institute 2015, 107, 569-578, 10.1093/jnci/dju428.
- Andrew G Renehan; Margaret Tyson; Matthias Egger; Richard F Heller; Marcel Zwahlen; Body-mass index and incidence of cancer: a systematic review and meta-analysis of prospective observational studies. The Lancet 2008, 371, 569-578, 10.1016/s0140-6736(08)60269-x.
- D. S. M. Chan; A. R. Vieira; D. Aune; E. V. Bandera; D. C. Greenwood; A. McTiernan; D. Navarro Rosenblatt; I. Thune; R. Vieira; T. Norat; et al. Body mass index and survival in women with breast cancer—systematic literature review and meta-analysis of 82 follow-up studies. Annals of Oncology 2014, 25, 1901-1914, 10.1093/annonc/mdu042.
- Wambui G Gathirua-Mwangi; Julie R Palmer; Victoria Champion; Nelsy Castro-Webb; Andrew C Stokes; Lucile Adams-Campbell; Andrew R Marley; Michele R Forman; Lynn Rosenberg; Kimberly A Bertrand; et al. Maximum and Time-Dependent Body Mass Index and Breast Cancer Incidence Among Postmenopausal Women in the Black Women’s Health Study. American Journal of Epidemiology 2022, 191, 646-654, 10.1093/aje/kwac004.
- Richa Chauhan; Vinita Trivedi; Rita Rani; Usha Singh; A comparative analysis of body mass index with estrogen receptor, progesterone receptor and human epidermal growth factor receptor 2 status in pre- and postmenopausal breast cancer patients. Journal of Mid-life Health 2019, 11, 210-216, 10.4103/jmh.jmh_97_20.
- Donjete Statovci; Mònica Aguilera; John Mac Sharry; Silvia Melgar; The Impact of Western Diet and Nutrients on the Microbiota and Immune Response at Mucosal Interfaces. Frontiers in Immunology 2017, 8, 838, 10.3389/fimmu.2017.00838.
- Cielo García-Montero; Oscar Fraile-Martínez; Ana Gómez-Lahoz; Leonel Pekarek; Alejandro Castellanos; Fernando Noguerales-Fraguas; Santiago Coca; Luis Guijarro; Natalio García-Honduvilla; Angel Asúnsolo; et al.Lara Sanchez-TrujilloGuillermo LaheraJulia BujanJorge MonserratMelchor Álvarez-MonMiguel Álvarez-MonMiguel Ortega Nutritional Components in Western Diet Versus Mediterranean Diet at the Gut Microbiota–Immune System Interplay. Implications for Health and Disease. Nutrients 2021, 13, 699, 10.3390/nu13020699.
- Marit Zinöcker; Inge Lindseth; The Western Diet–Microbiome-Host Interaction and Its Role in Metabolic Disease. Nutrients 2018, 10, 365, 10.3390/nu10030365.
- Megan Cully; Han You; Arnold J. Levine; Tak W. Mak; Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nature Reviews Cancer 2006, 6, 184-192, 10.1038/nrc1819.
- Elizabeth Haythorne; Maria Rohm; Martijn Van De Bunt; Melissa F. Brereton; Andrei I. Tarasov; Thomas S. Blacker; Gregor Sachse; Mariana Silva Dos Santos; Raul Terron Exposito; Simon Davis; et al.Otto BabaRoman FischerMichael R. DuchenPatrik RorsmanJames I. MacraeFrances M. Ashcroft Diabetes causes marked inhibition of mitochondrial metabolism in pancreatic β-cells. Nature Communications 2019, 10, 1-17, 10.1038/s41467-019-10189-x.
- Philip Newsholme; Kevin N. Keane; Rodrigo Carlessi; Vinicius Cruzat; Oxidative stress pathways in pancreatic β-cells and insulin-sensitive cells and tissues: importance to cell metabolism, function, and dysfunction. American Journal of Physiology-Cell Physiology 2019, 317, C420-C433, 10.1152/ajpcell.00141.2019.
- Rainer J Klement; Ulrike Kämmerer; Is there a role for carbohydrate restriction in the treatment and prevention of cancer?. Nutrition & Metabolism 2010, 8, 75-75, 10.1186/1743-7075-8-75.
- Christopher R. LaPensee; Eric R. Hugo; Nira Ben-Jonathan; Insulin Stimulates Interleukin-6 Expression and Release in LS14 Human Adipocytes through Multiple Signaling Pathways. Endocrinology 2008, 149, 5415-5422, 10.1210/en.2008-0549.
- T. Makino; Y. Noguchi; T. Yoshikawa; C. Doi; K. Nomura; Circulating interleukin 6 concentrations and insulin resistance in patients with cancer. British Journal of Surgery 1998, 85, 1658-1662, 10.1046/j.1365-2168.1998.00938.x.
- J L McCall; J A Tuckey; B R Parry; Serum tumour necrosis factor alpha and insulin resistance in gastrointestinal cancer. British Journal of Surgery 1992, 79, 1361-1363, 10.1002/bjs.1800791240.
- Mei Tian; Hong Zhang; Yoshiki Nakasone; Kenji Mogi; Keigo Endo; Expression of Glut-1 and Glut-3 in untreated oral squamous cell carcinoma compared with FDG accumulation in a PET study. European Journal of Pediatrics 2003, 31, 5-12, 10.1007/s00259-003-1316-9.
- Hui-Lu Zhang; Ming-Da Wang; Xu Zhou; Chen-Jie Qin; Gong-Bo Fu; Liang Tang; Han Wu; Shuai Huang; Ling-Hao Zhao; Min Zeng; et al.Jiao LiuDan CaoLin-Na GuoHong-Yang WangHe-Xin YanJie Liu Blocking preferential glucose uptake sensitizes liver tumor-initiating cells to glucose restriction and sorafenib treatment. Cancer Letters 2016, 388, 1-11, 10.1016/j.canlet.2016.11.023.
- Rylee Maldonado; Chloe Adrienna Talana; Cassaundra Song; Alyssa Dixon; Kahealani Uehara; Michael Weichhaus; β‑hydroxybutyrate does not alter the effects of glucose deprivation on breast cancer cells. Oncology Letters 2020, 21, 1-1, 10.3892/ol.2020.12326.
- Sharon Varghese; Samson Mathews Samuel; Elizabeth Varghese; Peter Kubatka; Dietrich Büsselberg; High Glucose Represses the Anti-Proliferative and Pro-Apoptotic Effect of Metformin in Triple Negative Breast Cancer Cells. Biomolecules 2019, 9, 16, 10.3390/biom9010016.
- Qianying Zuo; Ayca Nazli Mogol; Yu-Jeh Liu; Ashlie Santaliz Casiano; Christine Chien; Jenny Drnevich; Ozan Berk Imir; Eylem Kulkoyluoglu-Cotul; Nicole Hwajin Park; David J. Shapiro; et al.Ben Ho ParkYvonne ZieglerBenita S. KatzenellenbogenEvelyn ArandaJohn D. O'NeillAkshara Singareeka RaghavendraDebu TripathyZeynep Madak Erdogan Targeting Metabolic Adaptations in the Breast Cancer–Liver Metastatic Niche Using Dietary Approaches to Improve Endocrine Therapy Efficacy. Molecular Cancer Research 2022, 20, 923-937, 10.1158/1541-7786.mcr-21-0781.
- Marin E. Healy; Jenny D.Y. Chow; Frances L. Byrne; David S. Breen; Norbert Leitinger; Chien Li; Carolin Lackner; Stephen H. Caldwell; Kyle L. Hoehn; Dietary effects on liver tumor burden in mice treated with the hepatocellular carcinogen diethylnitrosamine. Journal of Hepatology 2014, 62, 599-606, 10.1016/j.jhep.2014.10.024.
- Lars P. Bechmann; Rebekka A. Hannivoort; Guido Gerken; Gökhan S. Hotamisligil; Michael Trauner; Ali Canbay; The interaction of hepatic lipid and glucose metabolism in liver diseases. Journal of Hepatology 2012, 56, 952-964, 10.1016/j.jhep.2011.08.025.
- Xing-Yu Zhong; Xiu-Ming Yuan; Ying-Ying Xu; Miao Yin; Wei-Wei Yan; Shao-Wu Zou; Li-Ming Wei; Hao-Jie Lu; Yi-Ping Wang; Qun-Ying Lei; et al. CARM1 Methylates GAPDH to Regulate Glucose Metabolism and Is Suppressed in Liver Cancer. Cell Reports 2018, 24, 3207-3223, 10.1016/j.celrep.2018.08.066.
- Jelena Krstic; Isabel Reinisch; Katharina Schindlmaier; Markus Galhuber; Zina Riahi; Natascha Berger; Nadja Kupper; Elisabeth Moyschewitz; Martina Auer; Helene Michenthaler; et al.Christoph NössingMaria R. DepaoliJeta Ramadani-MujaSinem UsluerSarah StryeckMartin PichlerBeate RinnerAlexander J. A. DeutschAndreas ReinischTobias MadlRiccardo Zenezini ChiozziAlbert J. R. HeckMeritxell HuchRoland MalliAndreas Prokesch Fasting improves therapeutic response in hepatocellular carcinoma through p53-dependent metabolic synergism. Science Advances 2022, 8, 2635, 10.1126/sciadv.abh2635.
- Wahdan Alaswad Rs; Edgerton Sm; Salem Hs; Thor Ad; Metformin Targets Glucose Metabolism in Triple Negative Breast Cancer. Journal of Oncology Translational Research 2017, 04, 1-6, 10.4172/2476-2261.1000129.
- Ruchi Roy; Eun‐Ryeong Hahm; Alexander G. White; Carolyn J. Anderson; Shivendra V. Singh; AKT‐dependent sugar addiction by benzyl isothiocyanate in breast cancer cells. Molecular Carcinogenesis 2019, 58, 996-1007, 10.1002/mc.22988.
- Udi Gluschnaider; Rachel Hertz; Sarit Ohayon; Elia Smeir; Martha Smets; Eli Pikarsky; Jacob Bar-Tana; Long-Chain Fatty Acid Analogues Suppress Breast Tumorigenesis and Progression. Cancer Research 2014, 74, 6991-7002, 10.1158/0008-5472.can-14-0385.
- Min Wei; Sebastian Brandhorst; Mahshid Shelehchi; Hamed Mirzaei; Chia Wei Cheng; Julia Budniak; Susan Groshen; Wendy J. Mack; Esra Guen; Stefano Di Biase; et al.Pinchas CohenTodd E. MorganTanya DorffKurt HongAndreas MichalsenAlessandro LavianoValter D. Longo Fasting-mimicking diet and markers/risk factors for aging, diabetes, cancer, and cardiovascular disease. Science Translational Medicine 2017, 9, 377, 10.1126/scitranslmed.aai8700.
- A.M. Poff; C. Ari; P. Arnold; T.N. Seyfried; D.P. D'Agostino; Ketone supplementation decreases tumor cell viability and prolongs survival of mice with metastatic cancer. International Journal of Cancer 2014, 135, 1711-1720, 10.1002/ijc.28809.
- Bryan G. Allen; Sudershan K. Bhatia; Carryn M. Anderson; Julie M. Eichenberger-Gilmore; Zita A. Sibenaller; Kranti A. Mapuskar; Joshua D. Schoenfeld; John M. Buatti; Douglas R. Spitz; Melissa A. Fath; et al. Ketogenic diets as an adjuvant cancer therapy: History and potential mechanism. Redox Biology 2014, 2, 963-970, 10.1016/j.redox.2014.08.002.
- P. Rigo; P. Paulus; B. J. Kaschten; R. Hustinx; T. Bury; G. Jerusalem; T. Benoit; J. Foidart-Willems; Oncological applications of positron emission tomography with fluorine-18 fluorodeoxyglucose. European Journal of Pediatrics 1996, 23, 1641-1674, 10.1007/bf01249629.
- Kosuke Kaji; Norihisa Nishimura; Kenichiro Seki; Shinya Sato; Soichiro Saikawa; Keisuke Nakanishi; Masanori Furukawa; Hideto Kawaratani; Mitsuteru Kitade; Kei Moriya; et al.Tadashi NamisakiHitoshi Yoshiji Sodium glucose cotransporter 2 inhibitor canagliflozin attenuates liver cancer cell growth and angiogenic activity by inhibiting glucose uptake. International Journal of Cancer 2017, 142, 1712-1722, 10.1002/ijc.31193.
- Xiao Zhang; Yongxia Qiao; Qi Wu; Yan Chen; Shaowu Zou; Xiangfan Liu; Guoqing Zhu; Yinghui Zhao; Yuxin Chen; Yongchun Yu; et al.Qiuhui PanJiayi WangFenyong Sun The essential role of YAP O-GlcNAcylation in high-glucose-stimulated liver tumorigenesis. Nature Communications 2017, 8, 15280, 10.1038/ncomms15280.
- Pengcheng Bu; Kai-Yuan Chen; Kun Xiang; Christelle Johnson; Scott B. Crown; Nikolai Rakhilin; Yiwei Ai; Lihua Wang; Rui Xi; Inna Astapova; et al.Yan HanJiahe LiBradley B. BarthMin LuZiyang GaoRobert MinesLiwen ZhangMark HermanDavid HsuGuo-Fang ZhangXiling Shen Aldolase B-Mediated Fructose Metabolism Drives Metabolic Reprogramming of Colon Cancer Liver Metastasis. Cell Metabolism 2018, 27, 1249-1262.e4, 10.1016/j.cmet.2018.04.003.
- Otto Warburg; On the Origin of Cancer Cells. Science 1956, 123, 309-314, 10.1126/science.123.3191.309.
- Nùkhet Aykin-Burns; Iman M. Ahmad; Yueming Zhu; Larry W. Oberley; Douglas R. Spitz; Increased levels of superoxide and H2O2 mediate the differential susceptibility of cancer cells versus normal cells to glucose deprivation. Biochemical Journal 2009, 418, 29-37, 10.1042/bj20081258.
- L.G. Boros; P.W.N. Lee; J.L. Brandes; Marta Cascante; Peter Muscarella; W.J. Schirmer; W.S. Melvin; E.C. Ellison; Nonoxidative pentose phosphate pathways and their direct role in ribose synthesis in tumors: is cancer a disease of cellular glucose metabolism?. Medical Hypotheses 1997, 50, 55-59, 10.1016/s0306-9877(98)90178-5.
- Daniela D. Weber; Sepideh Aminazdeh-Gohari; Barbara Kofler; Ketogenic diet in cancer therapy. Aging 2018, 10, 164-165, 10.18632/aging.101382.
- Douglas C Wallace; Mitochondria and cancer. Nature Reviews Cancer 2012, 12, 685-698, 10.1038/nrc3365.
- Garry R. Buettner; Superoxide Dismutase in Redox Biology: The Roles of Superoxide and Hydrogen Peroxide. Anti-Cancer Agents in Medicinal Chemistry 2011, 11, 341-346, 10.2174/187152011795677544.
- Richard L. Veech; Ketone ester effects on metabolism and transcription. Journal of Lipid Research 2014, 55, 2004-2006, 10.1194/jlr.r046292.
- Benjamin D. Hopkins; Chantal Pauli; Xing Du; Diana G. Wang; Xiang Li; David Wu; Solomon C. Amadiume; Marcus Goncalves; Cindy Hodakoski; Mark R. Lundquist; et al.Rohan BarejaYan MaEmily M. HarrisAndrea SbonerHimisha BeltranMark RubinSiddhartha MukherjeeLewis C. Cantley Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature 2018, 560, 499-503, 10.1038/s41586-018-0343-4.
- Victor W. Ho; Kelvin Leung; Anderson Hsu; Beryl Luk; June Lai; Sung Yuan Shen; Andrew I. Minchinton; Dawn Waterhouse; Marcel B. Bally; Wendy Lin; et al.Brad H. NelsonLaura M. SlyGerald Krystal A Low Carbohydrate, High Protein Diet Slows Tumor Growth and Prevents Cancer Initiation. Cancer Research 2011, 71, 4484-4493, 10.1158/0008-5472.can-10-3973.
- Matthew G. Vander Heiden; Lewis C. Cantley; Craig B. Thompson; Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029-1033, 10.1126/science.1160809.
- Frances L. Byrne; Stefan R. Hargett; Sujoy Lahiri; R. Jack Roy; Stuart S. Berr; Stephen H. Caldwell; Kyle L. Hoehn; Serial MRI Imaging Reveals Minimal Impact of Ketogenic Diet on Established Liver Tumor Growth. Cancers 2018, 10, 312, 10.3390/cancers10090312.
- Jocelyn Tan-Shalaby; Ketogenic Diets and Cancer: Emerging Evidence.. Federal practitioner : for the health care professionals of the VA, DoD, and PHS 2017, 34, 37S-42S.
- Bryan G. Allen; Sudershan K. Bhatia; John M. Buatti; Kristin E. Brandt; Kaleigh E. Lindholm; Anna M. Button; Luke I. Szweda; Brian J. Smith; Douglas R. Spitz; Melissa A. Fath; et al. Ketogenic Diets Enhance Oxidative Stress and Radio-Chemo-Therapy Responses in Lung Cancer Xenografts. Clinical Cancer Research 2013, 19, 3905-3913, 10.1158/1078-0432.ccr-12-0287.
- Mohammed G. Abdelwahab; Kathryn E. Fenton; Mark C. Preul; Jong M. Rho; Andrew Lynch; Phillip Stafford; Adrienne C. Scheck; The Ketogenic Diet Is an Effective Adjuvant to Radiation Therapy for the Treatment of Malignant Glioma. PLOS ONE 2012, 7, e36197, 10.1371/journal.pone.0036197.
- H. Bobby Fokidis; Mei Yieng Chin; Victor W. Ho; Hans H. Adomat; Kiran K. Soma; Ladan Fazli; Ka Mun Nip; Michael Cox; Gerald Krystal; Amina Zoubeidi; et al.Emma S. Tomlinson Guns A low carbohydrate, high protein diet suppresses intratumoral androgen synthesis and slows castration-resistant prostate tumor growth in mice. The Journal of Steroid Biochemistry and Molecular Biology 2015, 150, 35-45, 10.1016/j.jsbmb.2015.03.006.
- Victor W. Ho; Melisa J. Hamilton; Ngoc-Ha Thi Dang; Brian E. Hsu; Hans H. Adomat; Emma S. Guns; Aalim Weljie; Ismael Samudio; Kevin L. Bennewith; Gerald Krystal; et al. A low carbohydrate, high protein diet combined with celecoxib markedly reduces metastasis. Carcinogenesis 2014, 35, 2291-2299, 10.1093/carcin/bgu147.
- Regina T. Martuscello; Vinata Vedam-Mai; David J. McCarthy; Michael E. Schmoll; Musa A. Jundi; Christopher D. Louviere; Benjamin G. Griffith; Colby L. Skinner; Oleg Suslov; Loic P. Deleyrolle; et al.Brent A. Reynolds A Supplemented High-Fat Low-Carbohydrate Diet for the Treatment of Glioblastoma. Clinical Cancer Research 2016, 22, 2482-2495, 10.1158/1078-0432.ccr-15-0916.
- Irene Caffa; Vanessa Spagnolo; Claudio Vernieri; Francesca Valdemarin; Pamela Becherini; Min Wei; Sebastian Brandhorst; Chiara Zucal; Else Driehuis; Lorenzo Ferrando; et al.Francesco PiacenteAlberto TagliaficoMichele CilliLuca MastracciValerio G. VelloneSilvano PiazzaAnna Laura CremoniniRaffaella GradaschiCarolina ManteroMario PassalacquaAlberto BallestreroGabriele ZoppoliMichele CeaAnnalisa ArrighiPatrizio OdettiFiammetta MonacelliGiulia SalvadoriSalvatore CortellinoHans CleversFilippo De BraudSamir G. SukkarAlessandro ProvenzaniValter D. LongoAlessio Nencioni Fasting-mimicking diet and hormone therapy induce breast cancer regression. Nature 2020, 583, 620-624, 10.1038/s41586-020-2502-7.
- Rainer J. Klement; Colin Champ; Christoph Otto; Ulrike Kämmerer; Anti-Tumor Effects of Ketogenic Diets in Mice: A Meta-Analysis. PLOS ONE 2016, 11, e0155050-e0155050, 10.1371/journal.pone.0155050.
- Lifeng Yang; Tara TeSlaa; Serina Ng; Michel Nofal; Lin Wang; Taijin Lan; Xianfeng Zeng; Alexis Cowan; Matthew McBride; Wenyun Lu; et al.Shawn DavidsonGaoyang LiangTae Gyu OhMichael DownesRonald EvansDaniel Von HoffJessie Yanxiang GuoHaiyong HanJoshua D. Rabinowitz Ketogenic diet and chemotherapy combine to disrupt pancreatic cancer metabolism and growth. Med 2022, 3, 119-136.e8, 10.1016/j.medj.2021.12.008.
- Stefano Di Biase; Hong Seok Shim; Kyung Hwa Kim; Manlio Vinciguerra; Francesca Rappa; Min Wei; Sebastian Brandhorst; Francesco Cappello; Hamed Mirzaei; Changhan Lee; et al.Valter D. Longo Fasting regulates EGR1 and protects from glucose- and dexamethasone-dependent sensitization to chemotherapy. PLOS Biology 2017, 15, e2001951, 10.1371/journal.pbio.2001951.
- Giulia Salvadori; Federica Zanardi; Fabio Iannelli; Riccardo Lobefaro; Claudio Vernieri; Valter D. Longo; Fasting-mimicking diet blocks triple-negative breast cancer and cancer stem cell escape. Cell Metabolism 2021, 33, 2247-2259.e6, 10.1016/j.cmet.2021.10.008.
- Mohamed Elgendy; Marco Cirò; Amir Hosseini; Jakob Weiszmann; Luca Mazzarella; Elisa Ferrari; Riccardo Cazzoli; Giuseppe Curigliano; Andrea DeCensi; Bernardo Bonanni; et al.Alfredo BudillonPier Giuseppe PelicciVeerle JanssensManfred OgrisManuela BaccariniLuisa LanfranconeWolfram WeckwerthMarco FoianiSaverio Minucci Combination of Hypoglycemia and Metformin Impairs Tumor Metabolic Plasticity and Growth by Modulating the PP2A-GSK3β-MCL-1 Axis. Cancer Cell 2019, 35, 798-815.e5, 10.1016/j.ccell.2019.03.007.
- Patrycja Puchalska; Peter A. Crawford; Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metabolism 2017, 25, 262-284, 10.1016/j.cmet.2016.12.022.
- Lori Laffel; Ketone bodies: a review of physiology, pathophysiology and application of monitoring to diabetes. Diabetes/Metabolism Research and Reviews 1999, 15, 412-426, 10.1002/(sici)1520-7560(199911/12)15:6<412::aid-dmrr72>3.0.co;2-8.
- John C. Newman; Eric Verdin; β-Hydroxybutyrate: A Signaling Metabolite. Annual Review of Nutrition 2017, 37, 51-76, 10.1146/annurev-nutr-071816-064916.
- Dhillon, Kiranjit K.; Gupta, Sonu. Biochemistry, Ketogenesis; Stat Pearls Publishing: Treasure Island, FL, USA, 2022; pp. 1.
- Do Young Kim; Jong M Rho; The ketogenic diet and epilepsy. Current Opinion in Clinical Nutrition and Metabolic Care 2008, 11, 113-120, 10.1097/mco.0b013e3282f44c06.
- Yafei Duan; Yue Zhang; Hongbiao Dong; Yun Wang; Jiasong Zhang; Effects of dietary poly-β-hydroxybutyrate (PHB) on microbiota composition and the mTOR signaling pathway in the intestines of litopenaeus vannamei. Journal of Microbiology 2017, 55, 946-954, 10.1007/s12275-017-7273-y.
- Chao Huang; Peng Wang; Xing Xu; Yaru Zhang; Yu Gong; Wenfeng Hu; Minhui Gao; Yue Wu; Yong Ling; Xi Zhao; et al.Yibin QinRongrong YangWei Zhang The ketone body metabolite β-hydroxybutyrate induces an antidepression-associated ramification of microglia via HDACs inhibition-triggered Akt-small RhoGTPase activation. Glia 2017, 66, 256-278, 10.1002/glia.23241.
- Claudio Vernieri; Giovanni Fucà; Francesca Ligorio; Veronica Huber; Andrea Vingiani; Fabio Iannelli; Alessandra Raimondi; Darawan Rinchai; Gianmaria Frigè; Antonino Belfiore; et al.Luca LalliClaudia ChiodoniValeria CancilaFederica ZanardiArta AjaziSalvatore CortellinoViviana VallacchiPaola SquarcinaAgata CovaSamantha PescePaola FratiRaghvendra MallPaola Antonia CorsettoAngela Maria RizzoCristina FerrarisSecondo FolliMarina Chiara GarassinoGiuseppe CapriGiulia BianchiMario Paolo ColomboSaverio MinucciMarco FoianiValter Daniel LongoGiovanni ApoloneValter TorriGiancarlo PruneriDavide BedognettiLicia RivoltiniFilippo de Braud Fasting-Mimicking Diet Is Safe and Reshapes Metabolism and Antitumor Immunity in Patients with Cancer. Cancer Discovery 2021, 12, 90-107, 10.1158/2159-8290.cd-21-0030.
- Young-Min Han; Tharmarajan Ramprasath; Ming-Hui Zou; β-hydroxybutyrate and its metabolic effects on age-associated pathology. Experimental & Molecular Medicine 2020, 52, 548-555, 10.1038/s12276-020-0415-z.
- Dae Hyun Kim; Min Hi Park; Sugyeong Ha; Eun Jin Bang; Yujeong Lee; A Kyoung Lee; Jaewon Lee; Byung Pal Yu; Hae Young Chung; Anti-inflammatory action of β-hydroxybutyrate via modulation of PGC-1α and FoxO1, mimicking calorie restriction. Aging 2019, 11, 1283-1304, 10.18632/aging.101838.
- Tadahiro Shimazu; Matthew D. Hirschey; John Newman; Wenjuan He; Kotaro Shirakawa; Natacha Le Moan; Carrie A. Grueter; Hyungwook Lim; Laura R. Saunders; Robert D. Stevens; et al.Christopher B. NewgardRobert V. FareseRafael de CaboScott UlrichKaterina AkassoglouEric Verdin Suppression of Oxidative Stress by β-Hydroxybutyrate, an Endogenous Histone Deacetylase Inhibitor. Science 2013, 339, 211-214, 10.1126/science.1227166.
- Kishor Pant; Estanislao Peixoto; Seth Richard; Sergio A. Gradilone; Role of Histone Deacetylases in Carcinogenesis: Potential Role in Cholangiocarcinoma. Cells 2020, 9, 780, 10.3390/cells9030780.
- Ubaldo E. Martinez-Outschoorn; Marco Prisco; Adam Ertel; Aristotelis Tsirigos; Zhao Lin; Stephanos Pavlides; Chengwang Wang; Neal Flomenberg; Erik S. Knudsen; Anthony Howell; et al.Richard G. PestellFederica SotgiaMichael P Lisanti Ketones and lactate increase cancer cell “stemness,” driving recurrence, metastasis and poor clinical outcome in breast cancer. Cell Cycle 2011, 10, 1271-1286, 10.4161/cc.10.8.15330.
- Gloria Bonuccelli; Aristotelis Tsirigos; Diana Whitaker-Menezes; Stephanos Pavlides; Richard G. Pestell; Barbara Chiavarina; Philippe Frank; Neal Flomenberg; Anthony Howell; Ubaldo Martinez-Outschoorn; et al.Federica SotgiaMichael P. Lisanti Ketones and lactate “fuel” tumor growth and metastasis. Cell Cycle 2010, 9, 3506-3514, 10.4161/cc.9.17.12731.
- Chun-Kai Huang; Po-Hao Chang; Wen-Hung Kuo; Chi-Long Chen; Yung-Ming Jeng; King-Jen Chang; Jin-Yuh Shew; Chun-Mei Hu; Wen-Hwa Lee; Adipocytes promote malignant growth of breast tumours with monocarboxylate transporter 2 expression via β-hydroxybutyrate. Nature Communications 2017, 8, 14706, 10.1038/ncomms14706.
- Loreta M. Rodrigues; Santiago Uribe-Lewis; Basetti Madhu; Davina J. Honess; Marion Stubbs; John R. Griffiths; The action of β-hydroxybutyrate on the growth, metabolism and global histone H3 acetylation of spontaneous mouse mammary tumours: evidence of a β-hydroxybutyrate paradox. Cancer & Metabolism 2017, 5, 1-13, 10.1186/s40170-017-0166-z.
- Surendra K Shukla; Teklab Gebregiworgis; Vinee Purohit; Nina V Chaika; Venugopal Gunda; Prakash Radhakrishnan; Kamiya Mehla; Iraklis I Pipinos; Robert Powers; Fang Yu; et al.Pankaj K Singh Metabolic reprogramming induced by ketone bodies diminishes pancreatic cancer cachexia. Cancer & Metabolism 2014, 2, 18-18, 10.1186/2049-3002-2-18.
- Daisuke Mikami; Mamiko Kobayashi; Junsuke Uwada; Takashi Yazawa; Kazuko Kamiyama; Kazuhisa Nishimori; Yudai Nishikawa; Sho Nishikawa; Seiji Yokoi; Takanobu Taniguchi; et al.Masayuki Iwano β-Hydroxybutyrate enhances the cytotoxic effect of cisplatin via the inhibition of HDAC/survivin axis in human hepatocellular carcinoma cells. Journal of Pharmacological Sciences 2019, 142, 1-8, 10.1016/j.jphs.2019.10.007.
- Melanie Schmidt; Nadja Pfetzer; Micheal Schwab; Ingrid Strauss; Ulrike Kämmerer; Effects of a ketogenic diet on the quality of life in 16 patients with advanced cancer: A pilot trial. Nutrition & Metabolism 2011, 8, 54-54, 10.1186/1743-7075-8-54.
- Catharina Bartmann; Sudha R. Janaki Raman; Jessica Flöter; Almut Schulze; Katrin Bahlke; Jana Willingstorfer; Maria Strunz; Achim Wöckel; Rainer J. Klement; Michaela Kapp; et al.Cholpon S. DjuzenovaChristoph OttoUlrike Kämmerer Beta-hydroxybutyrate (3-OHB) can influence the energetic phenotype of breast cancer cells, but does not impact their proliferation and the response to chemotherapy or radiation. Cancer & Metabolism 2018, 6, 8, 10.1186/s40170-018-0180-9.
- Dallas R. Donohoe; Leonard B. Collins; Aminah Wali; Rebecca Bigler; Wei Sun; Scott J. Bultman; The Warburg Effect Dictates the Mechanism of Butyrate-Mediated Histone Acetylation and Cell Proliferation. Molecular Cell 2012, 48, 612-626, 10.1016/j.molcel.2012.08.033.
- Joanne R. Lupton; Microbial Degradation Products Influence Colon Cancer Risk: the Butyrate Controversy. The Journal of Nutrition 2004, 134, 479-482, 10.1093/jn/134.2.479.
- Shunli Luo; Ziyin Li; Lianzhi Mao; Siqiang Chen; Suxia Sun; Sodium butyrate induces autophagy in colorectal cancer cells through LKB1/AMPK signaling. Journal of Physiology and Biochemistry 2018, 75, 53-63, 10.1007/s13105-018-0651-z.
- Vahid Salimi; Zahra Shahsavari; Banafsheh Safizadeh; Ameinh Hosseini; Narges Khademian; Masoumeh Tavakoli-Yaraki; Sodium butyrate promotes apoptosis in breast cancer cells through reactive oxygen species (ROS) formation and mitochondrial impairment. Lipids in Health and Disease 2017, 16, 1-11, 10.1186/s12944-017-0593-4.
- Claudio Vernieri; Francesca Ligorio; Emma Zattarin; Licia Rivoltini; Filippo De Braud; Fasting-mimicking diet plus chemotherapy in breast cancer treatment. Nature Communications 2020, 11, 1-4, 10.1038/s41467-020-18194-1.
- Stefanie de Groot; Rieneke T. Lugtenberg; Danielle Cohen; Marij J. P. Welters; Ilina Ehsan; Maaike P. G. Vreeswijk; Vincent T. H. B. M. Smit; Hiltje de Graaf; Joan B. Heijns; Johanneke E. A. Portielje; et al.Agnes J. van de WouwAlex L. T. ImholzLonneke W. KesselsSuzan VrijaldenhovenArnold BaarsElma Meershoek-Klein KranenbargMarjolijn Duijm-De CarpentierHein PutterJacobus J. M. van der HoevenJohan W. R. NortierValter D. LongoHanno PijlJudith R. KroepEmine GökerAnke J. M. PasAafke H. HonkoopA. Elise van Leeuwen-StokDutch Breast Cancer Research Group (BOOG) Fasting mimicking diet as an adjunct to neoadjuvant chemotherapy for breast cancer in the multicentre randomized phase 2 DIRECT trial. Nature Communications 2020, 11, 1-9, 10.1038/s41467-020-16138-3.
- Stefano Di Biase; Changhan Lee; Sebastian Brandhorst; Brianna Manes; Roberta Buono; Chia-Wei Cheng; Mafalda Cacciottolo; Alejandro Martin-Montalvo; Rafael de Cabo; Min Wei; et al.Todd E. MorganValter D. Longo Fasting-Mimicking Diet Reduces HO-1 to Promote T Cell-Mediated Tumor Cytotoxicity. Cancer Cell 2016, 30, 136-146, 10.1016/j.ccell.2016.06.005.