Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 4 by Jessie Wu and Version 3 by Jessie Wu.
Neuropathic pain is a chronic pain caused by tissue injury or disease involving the somatosensory nervous system, which seriously affects the patient’s body function and quality of life. Saponins are a class of compounds with diverse structures, consisting of sapogenin and glycosyl groups. The common ones of the saccharides that make up saponins are D-glucose, D-galactose, D-xylose, L-arabinose, and L-rhamnose, etc.
- neuropathic pain
- saponins
- mechanism
1. Ginsenosides
Ginsenosides are the major biologically active components of Ginseng, which have a wide range of pharmacological activities. According to the skeleton of their aglycones, ginsenosides can be classified into two groups, tetracyclic triterpene dammarane-type saponins (protopanaxadiol (PPD)-, protopanaxatriol (PPT)-type) (Figure 1A) and tetracyclic triterpene oleanane-type saponins [1][2][3]. So far, more than 100 different ginsenoside monomers have been isolated, such as ginsenosides Rb1, Rb2, Rc, Rd, Re, Rg1, and Rf, the pharmacological and pharmacokinetic properties of which are different [4][5].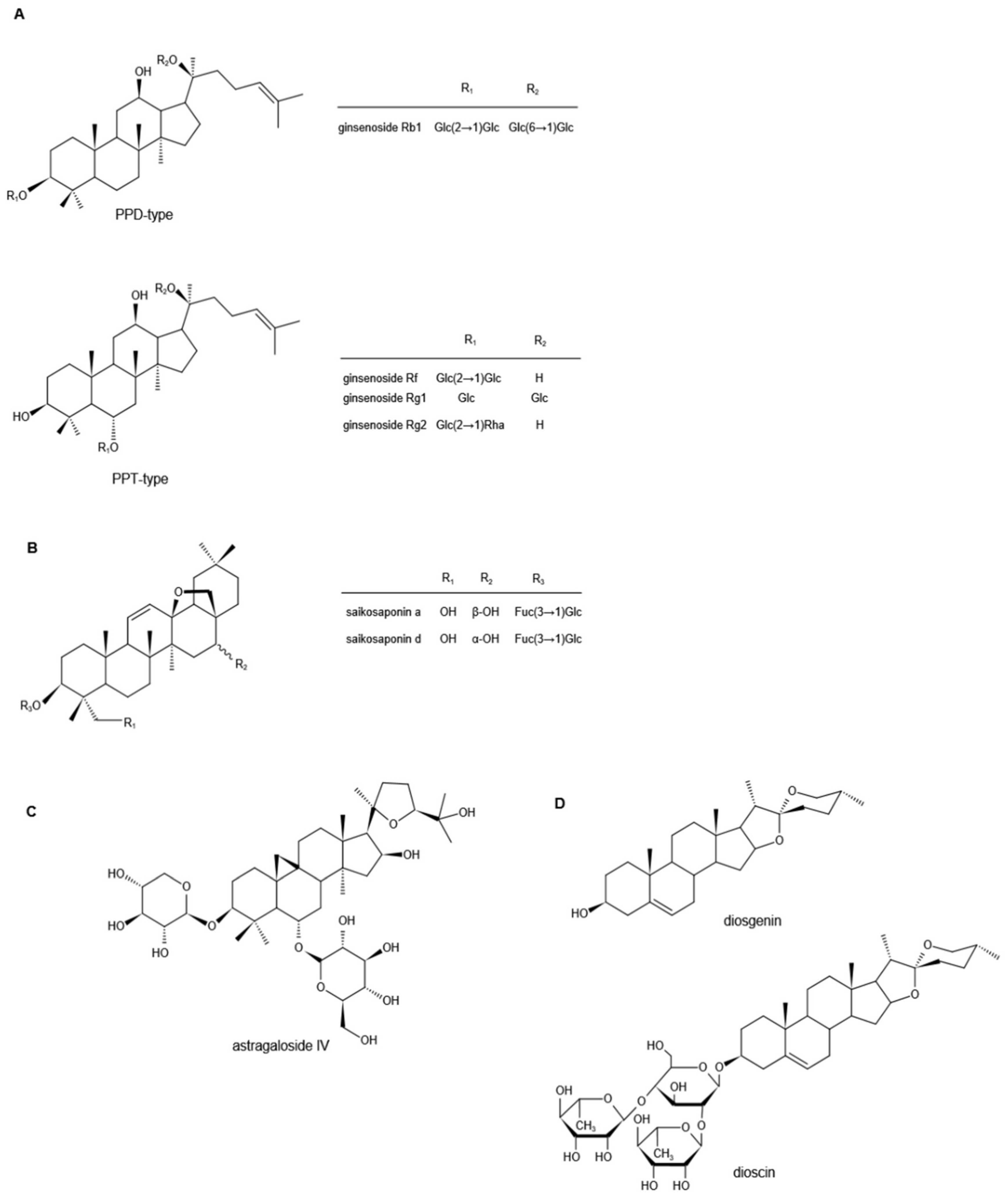
Figure 1. Chemical structures of ginsenosides (A), saikosaponins (B), astragaloside IV (C), and diosgenin and dioscin (D) (Note: PPD, protopanaxadiol; PPT, protopanaxatriol; Glc, glucoside; Rha, rhamnoside; Fuc, fructoside).
2. Saikosaponins
Saikosaponins are derived from Bupleurum or Bupleurum scorzonerifolium in the Umbelliferae, one of the traditional Chinese herbal medicines, and are the main active ingredients of Bupleurum [17]. So far, more than 100 kinds of saikosaponins have been isolated from Bupleurum, the main ones of which are oleanane and ursolic pentacyclic triterpene saponins [18][19][20][21][22]. According to their chemical structure, saikosaponins are divided into -A, -B, -C, -D, -M, -N, -P, and -T categories, and Saikosaponin D (SSD) is considered to be the most active one, followed by Saikosaponin A (SSA) [23][24]. Their chemical structures are shown in Figure 1B. Both in vivo and in vitro experimental studies have shown that saikosaponins can inhibit the activation of transient receptor potential ankyrin 1 (TRPA1) and significantly reduce the nociceptive response of animals induced by allyl isothiocyanate (AITC) [25]. Molecular docking and site-directed mutagenesis analyses demonstrated that saikosaponins bind to the TRPA1 hydrophobic pocket near the Asn855 residue, which once mutated to Ser and was previously united with enhanced pain perception in humans [25][26]. Gyeongbeen also reported that multiple administrations of SSD could significantly relieve mechanical hypersensitivity induced by vincristine, which was carried out partially by suppressing the activity of TRPA1 [25]. Therefore, it can be further speculated that SSD might play a certain therapeutic role in the neuropathic pain that is induced by chemotherapeutics, diabetes, or CCI, in which the expression and sensitivity of TRPA1 were changed as well, resulting in abnormal pain response and perception [27][28][29][30][31]. However, the analgesic effect of SSD is different between streptozotocin (STZ)- and paclitaxel-induced pain models. Short-term oral administration was effective in the former, while multiple administrations were required for the pain relief of the latter [32]. This indicates that the analgesic effect of SSD may not only act as an antagonist of TRPA1, but also exert anti-inflammatory activity to reduce the oxidative stress caused by nerve damage. Related research reported that SSD could restrain the translocation of the glucocorticoid receptor to the mitochondria, and decrease the H2O2-induced phosphorylation of extracellular-regulated kinase (ERK), c-Jun N-terminal kinase (c-JNK), and p38MAPK to downregulate the activity of neuronal PC12 cells [33][34][35]. It is well known that activation of NF-κB in both DRG and spinal cord neurons is associated with the transduction and processing of nociceptive messages. Therefore, inhibition of NF-κB can alleviate chronic painful states [36]. Studies have shown that SSA alleviates neuropathic pain by inhibiting CCI-induced elevation of p-p38 MAPK and NF-κB levels in the spinal cord [37]. In addition, cytokine dysregulation is one of the characteristic manifestations of neuropathic pain symptoms [38]. It could also be found that SSA significantly inhibited the expression of certain immune-related cytotoxic factors, including COX-2 and iNOS, and, likewise, the pro-inflammatory cytokines, such as TNF-α, IL-1β, and IL-6. Meanwhile, the expression of the important anti-inflammatory cytokine IL-10 was significantly upregulated, suggesting that it had anti-inflammatory activity in lipopolysaccharide (LPS)-stimulated macrophages [39][40]. Further research showed that SSA blocked the NF-κB signaling pathway by preventing phosphorylation of the NF-κB inhibitor α (IκBα), thereby allowing p65 NF-κB to remain in the cytoplasm, preventing it from translocating to the nucleus. In addition, SSA inhibited the MAPK signaling pathway by downregulating the phosphorylation of p38 MAPK, c-JNK, and ERK to exert the anti-inflammatory activity [40]. On the basis, SSA appeared to counteract the neurological function deficits after traumatic brain injury via inbiting aquaporin-4 (AQP-4) and matrix metalloprotein-9 (MMP-9) to account for its neuroprotective effects [41]. On the other hand, a study of Seong Shoon Yoon et al. expressed that SSA exhibited a significant inhibitory effect on morphine-reinforced behavior and drug addiction predominantly via mediating GABAB receptors [42][43]. Davoud Ahmadimoghaddam et al. reported that Bupleurum falcatum L. roots essential oil, of which SSA was one of the main constituents [44], exerted its antinociceptive and antiallodynic effects through the regulation of L-arginine-NO-cGMP-KATP channel pathways, as well as interaction with opioid, peroxisome proliferator-activated, and cannabinoid receptors [44]. The voltage-gated sodium channel Nav1.7 is a tetrodotoxin-sensitive sodium channel subtype and is encoded by SCN9A. It is well known that the dysfunction of Nav1.7 has the correlation with pain disorders [45]. Relevant research showed that SSA displayed the analgesic effects on the thermal pain and formalin-induced pain in mice via strong inhibitory effect on the peak currents of Nav1.7 [46]. The above studies have shown that SSD and SSA can exert analgesic effects in different neuropathic pain models through multiple pathways, and their mechanisms of action have similarities and differences. In the follow-up, weauthors can combine their structural characteristics with the mechanisms of action for deep analysis to provide a research basis for the precise regulation of the targets.3. Astragalosides
Astragali Radix, the dried roots of Astragalus membranaceus (Fisch.) Bge. var. mongholicus (Bge.) Hsiao, or Astragalus membranaceus (Fisch.) Bge., is known as a high-grade traditional Chinese medicine [47]. There are three main types of compounds in astragalus: saponins, flavonoids, and polysaccharides, and triterpene saponins are the major constituents [48][49][50]. It is reported that more than 40 kinds of saponins have been isolated and identified from the dried astragalus roots via HPLC and GC-MS, such as astragalosides I–VIII, acetylastragaloside, isoastragaloside I, III, astramembrannin II, cycloastragenol, cycloascauloside B, brachyoside B, astrasieversianin X, etc. [51][52][53][54]. Among these, astragaloside IV (AS-IV) is known as the major active ingredient and qualitative control biomarker. AS-IV is 3-O-beta-d-xylopyranosyl-6-O-beta-d-glucopyranosyl-cycloastragenol (Figure 1C), the molecular formula is C41H68O14 [55]. It is generally accepted that the transient receptor potential vanilloid 1 (TRPV1) channel is a polymodal receptor for various stimuli such as noxious heat and capsaicin, and is also an important pain sensor [56][57]. TRPV1 is overexpressed in Aδ fibers and C fibers in the situation of inflammation or nerve injury [58]. In addition, purinergic P2 × 3 receptors are ligand-gated nonselective cation channels, highly selectively expressed in small-diameter and medium-diameter sensory neurons related to nociceptive information, and play a key role in the generation and maintenance of pathological pain [59][60]. In the research by Guo-Bing Shi et al., AS-IV not only dramatically downregulated the expression of TRPV1 in Aδ fibers to remarkably upregulate the nociceptive threshold, but also inhibited P2 × 3 expression in DRG neurons to attenuate the mechanical allodynia [61]. Meanwhile, AS-IV restored the histological structure of the damaged sciatic nerve by accumulating glial cell-derived neurotrophic factor family receptorα1 (GFRα1), the glial cell derived neurotrophic factor (GDNF) selective receptor, in the debris of myelin between the Schwann cells and the damaged axon [61]. It also reduced the levels of GFRα1 and GDNF in DRG, which were highly expressed and induced by CCI, contributing to the restoration of injured nerve fibers [62][63]. In the peripheral nervous system, the appropriate dose of AS-IV could also greatly promote the regeneration of peripheral nerves [64]. Growth-associated protein 43 is lower in spinal cord segments L4–6 but active in growing neuronal axons in normal Balb/c mice. As a particular biomarker in nerve injury, it plays a vital role in nerve growth, and strongly associates with neuronal axon growth [55][65][66][67]. Previous research showed that AS-IV significantly upregulated the expression of growth-related protein 43 in regenerated nerve tissue, thereby increasing the number and diameter of myelinated nerve fibers in the sciatic nerve of mice, while elevating motor nerve conduction velocity and action potential amplitude [65]. Moreover, AS-IV also conducted analgesic effects on peripheral neuropathy in STZ-induced diabetic rats. Firstly, it reduced blood glucose and glycosylated hemoglobin (HbA1C) levels, and increased plasma insulin levels in diabetic rats [68]. It is crucial to control the levels of HbA1C because its concentration is closely related to the incidence of diabetes-related complications, which has been proven by clinical trials [69]. Secondly, AS-IV enhanced the activity of glutathione peroxidase in nerves, suppressed the activation of aldose reductase in erythrocytes, and decreased the accumulation of advanced glycation end products in both nerves and erythrocytes, which might not only activate the cellular antioxidant defense system, but also aggrandize the ability of antioxidative stress injury on peripheral nerves. Thirdly, AS-IV acted as the AR inhibitor, and then enhanced Na+, K+-ATPase activity, improved the delayed motor nerve conduction velocity, increased nerve blood flow, and prevented structural nerve fiber damage to correct peripheral nerve defects [68].4. Diosgenin
Diosgenin is a naturally occurring steroidal sapogenin and is abundant in nature. Primary sources of diosgenin include the three Dioscorea species and one Heterosmilax species, namely, D. zingiberensis, D. septemloba, D. collettii, and H. yunnanensis [70]. Diosgenin can also be obtained from fenugreek (T. foenum graecum Linn) and Costus speciosus [71][72][73]. It is a C27 spiroketal steroid sapogenin, 3β-hydroxy-5-spirostene (Figure 1D), and its molecular formula is C27H42O3 [74]. As a representational phytosteroid, diosgenin is an important basic raw material for the production of steroid hormone drugs and has received increasing attention in the pharmaceutical industry for decades [75]. In addition, diosgenin itself has a wide range of biological effects. The following studies mainly describe its role in neuropathic pain. Neuropathic pain, one of the common complications of diabetes mellitus, manifests as increased sensitivity to noxious stimuli [76]. To evaluate the effects of diosgenin in the treatment of diabetes-induced neuropathic pain, an in vivo study was performed on a rat model of STZ-induced diabetes. It was demonstrated that diosgenin upturned mechanical and thermal nociceptive thresholds and lowered pain scores at the late phase of the formalin test in diabetic rats [77]. Since elevated oxidative stress is one of the key factors in diabetes-related neurological dysfunction, it can lead to vascular dysfunction, resulting in intraneural hypoxia, which can lead to impaired motor and sensory nerve function [78][79]. Studies showed that diosgenin could reduce the content of malondialdehyde (MDA) in serum, DRG, and sciatic nerve of diabetic rats and restored the activities of superoxide dismutase (SOD) and catalase, thereby inhibiting oxidative stress and enhancing the function of the antioxidant defense system [77]. Furthermore, NF-κB, an important nuclear transcription factor, is responsible for the control of genes encoding inflammation and nociception-related mediators [80]. Upregulation of NF-κB in the DRG neurons of diabetic rats has been proven, and its inhibition significantly reduces nociceptive responses [81][82][83]. It reported that diosgenin downregulated the NF-κB p65/p50 signaling pathway in the LPS-induced lung injury model [84]. However, based on the available reports, there is no specific experimental research regarding whether diosgenin exerts its analgesic effect in diabetes-induced neuropathic pain by regulating NF-κB, and related research needs to be further developed. Nerve growth factor (NGF), as a neurotrophic factor, is a protein factor that plays a vital role in the maintenance of the growth, development, and function of sympathetic and sensory neurons. It stimulates the axon growth, maintains the axon size, prevents the postinjury death of mature neurons, and regulates various functions of the nervous system, including synaptic plasticity and neurotransmission [85][86]. In diabetic neuropathy, the function of NGF is impaired and the expression of NGF-related genes is modified, which are important factors in the progress of diabetic neuropathic pain. A study conducted by Tong Ho KANG et al. revealed that diosgenin upregulated the level of NGF in the sciatic nerve of diabetic rats. The comparable effects also reported that diosgenin increased the neurite outgrowth of PC12 cells, enhanced the sciatic nerve conduction velocity of diabetic mice by inducing NGF, reduced myelin disturbance, increased the area of myelinated axons, and improved the signal transmission of damaged axons, thereby alleviating diabetic neuropathic pain [87]. In addition to the diabetic neuropathy model, the role of diosgenin in the treatment of neuropathic pain has also been reported in the CCI rat model. In 2017, Wei-Xin Zhao et al. performed an in vivo study, and the results demonstrated that diosgenin could upregulate CCI-reduced mechanical withdrawal threshold and thermal withdrawal latency. This was due to the fact that diosgenin not only inhibited CCI-induced elevation of proinflammatory cytokines TNF-α, IL-1β and IL-2, but also suppressed oxidative stress in the spinal cord. Moreover, diosgenin remarkably restrained the expression of p-p38 MAPK and NF-κB in the spinal cord and eased neuropathic pain in CCI rats by inhibiting the activation of p38 MAPK and NF-κB signaling pathways [88]. In other research [89], sciatic-crushed-nerve injury in rats decreased the sciatic function index, which was widely used to evaluate functional gait [90], increased the c-Fos expression in the ventrolateral periaqueductal gray and paraventricular nucleus, restrained recovery of locomotor function caused by the overexpression of BDNF, and aggrandized expressions of COX-2 and iNOS that responded to inflammation. Fortunately, diosgenin was able to significantly improve the above pathological states, and exploited potential abilities in pain control and functional recovery after peripheral nerve injury.5. Saponin-Rich Extracts of O. sanctum
In addition to the analgesic effects of the above four plant saponins that have been identified with clear structures, saponin-rich extracts of O. sanctum have also been found with similar effects. O. sanctum is the aerial part of Ocimum basilicum, a plant of the Labiatae family. Modern pharmacological studies have illustrated that the chemical composition of O. sanctum is complex and the types are diverse, including volatile oils, flavonoids and their glycosides, coumarins, phenylpropanoids, and fatty acids, mainly volatile oils and flavonoids and their glycosides [91]. In addition, a variety of saponins have been isolated from the alcoholic extract of O. sanctum [92], the most important of which are pentacyclic triterpenoid saponins that are dominated by ursolic and oleanolic acids [93][94][95], and have a wide range of pharmacological effects. Oxidative stress [96] and alterations in calcium homeostasis [97] are thought to be closely associated with neuropathic pain. During neurological disorders, dysfunction of the intracellular calcium regulatory system produces oxidative stress [98], and increases in free radicals lead to neuronal degeneration and apoptosis. On the other hand, metabolic abnormalities [99], formation of protein aggregates [100], and changes in membrane permeability [101] caused by oxidative stress all increase calcium levels, and they act together to promote the deterioration of neuropathic pain. O. sanctum has a good antioxidant effect [102][103], protects against free radical damage [104], and is able to reduce calcium levels [105]. O. sanctum is used as a neurotonic in parts of India for the relief of headache, joint pain, and muscle pain. In the experiments conducted by Muthuraman et al., the administration of O. sanctum attenuated sciatic nerve transection-induced peripheral neuropathy and motor in-co-ordination, attenuated the amputation-induced reduction in thiobarbituric acid reactive species, total calcium, and glutathione levels in a dose-dependent manner [105]. It suggested that the analgesic effect of O. sanctum might be related to its antioxidation and reduction of calcium levels. Additionally, in other studies, treatment with O. sanctum and its saponin-rich fraction reduced neuropathic pain caused by chronic constrictive injury and chemotherapeutic agent vincristine, associated with its effects on the oxidative stress and calcium levels [105][106]. Based on the above findings, it can be observed that the downregulation of calcium levels by O. sanctum administration may be due to a direct effect on or secondary to a decrease in oxidative stress. Then, it produces an antinociceptive or antiapoptotic effect on neurons. It has been reported that Saponins have antioxidant [107] and calcium lowering effects [108]. Thus, the antinociceptive effect of O. sanctum saponins may be constructed through direct or indirect reduction of calcium levels. There is also evidence that O. sanctum leaves and seeds reduce uric acid levels in rabbits [109], and elevated uric acid levels are associated with gouty arthritis and other joint inflammation [110]. The ethanolic extract of O. sanctum can be antinociceptive, and involves the interaction of neurotransmitter systems such as opioid receptors and norepinephrine [111]. These studies support the traditional use of O. sanctum for the treatment of inflammation and pain, without excluding the effects of other active ingredients such as flavonoids and phenols.References
- Christensen, L.P. Ginsenosides chemistry, biosynthesis, analysis, and potential health effects. Adv. Food Nutr. Res. 2009, 55, 1–99.
- Zheng, M.; Xin, Y.; Li, Y.; Xu, F.; Xi, X.; Guo, H.; Cui, X.; Cao, H.; Zhang, X.; Han, C. Ginsenosides: A Potential Neuroprotective Agent. BioMed Res. Int. 2018, 2018, 8174345.
- Hou, M.; Wang, R.; Zhao, S.; Wang, Z. Ginsenosides in Panax genus and their biosynthesis. Acta Pharm. Sin. B 2021, 11, 1813–1834.
- Lee, H.S.; Lee, H.J.; Yu, H.J.; Ju do, W.; Kim, Y.; Kim, C.T.; Kim, C.J.; Cho, Y.J.; Kim, N.; Choi, S.Y.; et al. A comparison between high hydrostatic pressure extraction and heat extraction of ginsenosides from ginseng (Panax ginseng CA Meyer). J. Sci. Food Agric. 2011, 91, 1466–1473.
- Nag, S.A.; Qin, J.J.; Wang, W.; Wang, M.H.; Wang, H.; Zhang, R. Ginsenosides as Anticancer Agents: In vitro and in vivo Activities, Structure-Activity Relationships, and Molecular Mechanisms of Action. Front. Pharmacol. 2012, 3, 25.
- Lee, J.Y.; Choi, H.Y.; Park, C.S.; Kim, D.H.; Yune, T.Y. Total saponin extract, ginsenoside Rb1, and compound K alleviate peripheral and central neuropathic pain through estrogen receptors on rats. Phytother. Res. 2021, 35, 2119–2132.
- Gao, C.; Guo, X.; Weng, L.; Zhou, W.; Shen, Y.; Yu, Y.; Han, Y. Effect of ginsenoside Rg1 on activity of spinal microglia and expression of p38 mitogen-activated protein kinase/nuclear transcription factor-kappa B in neuropathic pain rats. Int. J. Anesth. Resus 2017, 38, 1084.
- Li, Y.; Chen, C.; Li, S.; Jiang, C. Ginsenoside Rf relieves mechanical hypersensitivity, depression-like behavior, and inflammatory reactions in chronic constriction injury rats. Phytother. Res. 2019, 33, 1095–1103.
- Wolf, G.; Yirmiya, R.; Goshen, I.; Iverfeldt, K.; Holmlund, L.; Takeda, K.; Shavit, Y. Impairment of interleukin-1 (IL-1) signaling reduces basal pain sensitivity in mice: Genetic, pharmacological and developmental aspects. Pain 2003, 104, 471–480.
- Miyoshi, K.; Obata, K.; Kondo, T.; Okamura, H.; Noguchi, K. Interleukin-18-mediated microglia/astrocyte interaction in the spinal cord enhances neuropathic pain processing after nerve injury. J. Neurosci. 2008, 28, 12775–12787.
- Ji, R.R.; Chamessian, A.; Zhang, Y.Q. Pain regulation by non-neuronal cells and inflammation. Science 2016, 354, 572–577.
- Sommer, C.; Leinders, M.; Üçeyler, N. Inflammation in the pathophysiology of neuropathic pain. Pain 2018, 159, 595–602.
- Huang, F.; Li, Y.N.; Yin, F.; Wu, Y.T.; Zhao, D.X.; Li, Y.; Zhang, Y.F.; Zhu, Q.S. Ginsenoside Rb1 inhibits neuronal apoptosis and damage, enhances spinal aquaporin 4 expression and improves neurological deficits in rats with spinal cord ischemiareperfusion injury. Mol. Med. Rep. 2015, 11, 3565–3572.
- Gao, X.-Q.; Yang, C.-X.; Chen, G.-J.; Wang, G.-Y.; Chen, B.; Tan, S.-K.; Liu, J.; Yuan, Q.-L. Ginsenoside Rb1 regulates the expressions of brain-derived neurotrophic factor and caspase-3 and induces neurogenesis in rats with experimental cerebral ischemia. J. Ethnopharmacol. 2010, 132, 393–399.
- Adamo, D.; Calabria, E.; Coppola, N.; Pecoraro, G.; Mignogna, M.D. Vortioxetine as a new frontier in the treatment of chronic neuropathic pain: A review and update. Ther. Adv. Psychopharmacol. 2021, 11, 1–19.
- Zhang, Q.L.; Li, S.Y.; Li, P. Effects of ginsenoside-Rg2 on mechanical allodynia, heat hyperalgeia, depressive state of rats with chronic sciatic nerve constriction injury. Zhongguo Ying Yong Sheng Li Xue Za Zhi 2019, 35, 228–231.
- Ruizhen, C.; Chunyan, Z. Research progress on pharmacological activities of Saikosaponins from Radix Bupleurum. Occup. Health 2021, 37, 568–576.
- He, Y.; Hu, Z.; Li, A.; Zhu, Z.; Yang, N.; Ying, Z.; He, J.; Wang, C.; Yin, S.; Cheng, S. Recent Advances in Biotransformation of Saponins. Molecules 2019, 24, 2365.
- Li, X.; Li, X.; Huang, N.; Liu, R.; Sun, R. A comprehensive review and perspectives on pharmacology and toxicology of saikosaponins. Phytomedicine 2018, 50, 73–87.
- Ashour, M.L.; Wink, M. Genus Bupleurum: A review of its phytochemistry, pharmacology and modes of action. J. Pharm. Pharm. 2011, 63, 305–321.
- Ebata, N.; Nakajima, K.; Hayashi, K.; Okada, M.; Maruno, M. Saponins from the root of Bupleurum falcatum. Phytochemistry 1996, 41, 895–901.
- Li, X.Q.; Song, Y.N.; Wang, S.J.; Rahman, K.; Zhu, J.Y.; Zhang, H. Saikosaponins: A review of pharmacological effects. J. Asian Nat. Prod. Res. 2018, 20, 399–411.
- Wong, V.K.; Zhou, H.; Cheung, S.S.; Li, T.; Liu, L. Mechanistic study of saikosaponin-d (Ssd) on suppression of murine T lymphocyte activation. J Cell Biochem. 2009, 107, 303–315.
- Wang, Y.L.; He, S.X.; Luo, J.Y. Progress in research on antitumor activity of saikosaponin and its mechanism. J. Chin. Integr. Med. 2006, 4, 98–100.
- Lee, G.; Choi, J.; Nam, Y.J.; Song, M.J.; Kim, J.K.; Kim, W.J.; Kim, P.; Lee, J.S.; Kim, S.; No, K.T.; et al. Identification and characterization of saikosaponins as antagonists of transient receptor potential A1 channel. Phytother. Res. 2020, 34, 788–795.
- Gupta, R.; Saito, S.; Mori, Y.; Itoh, S.G.; Okumura, H.; Tominaga, M. Structural basis of TRPA1 inhibition by HC-030031 utilizing species-specific differences. Sci. Rep. 2016, 6, 37460.
- Wang, S.; Kobayashi, K.; Kogure, Y.; Yamanaka, H.; Yamamoto, S.; Yagi, H.; Noguchi, K.; Dai, Y. Negative Regulation of TRPA1 by AMPK in Primary Sensory Neurons as a Potential Mechanism of Painful Diabetic Neuropathy. Diabetes 2018, 67, 98–109.
- Nativi, C.; Gualdani, R.; Dragoni, E.; Di Cesare Mannelli, L.; Sostegni, S.; Norcini, M.; Gabrielli, G.; la Marca, G.; Richichi, B.; Francesconi, O.; et al. A TRPA1 antagonist reverts oxaliplatin-induced neuropathic pain. Sci. Rep. 2013, 3, 2005.
- Nassini, R.; Gees, M.; Harrison, S.; De Siena, G.; Materazzi, S.; Moretto, N.; Failli, P.; Preti, D.; Marchetti, N.; Cavazzini, A.; et al. Oxaliplatin elicits mechanical and cold allodynia in rodents via TRPA1 receptor stimulation. Pain 2011, 152, 1621–1631.
- Descoeur, J.; Pereira, V.; Pizzoccaro, A.; Francois, A.; Ling, B.; Maffre, V.; Couette, B.; Busserolles, J.; Courteix, C.; Noel, J.; et al. Oxaliplatin-induced cold hypersensitivity is due to remodelling of ion channel expression in nociceptors. EMBO Mol. Med. 2011, 3, 266–278.
- Anderson, D.W.; Bradbury, K.A.; Schneider, J.S. Broad neuroprotective profile of nicotinamide in different mouse models of MPTP-induced parkinsonism. Eur. J. Neurosci. 2008, 28, 610–617.
- Lee, G.; Nam, Y.-J.; Kim, W.J.; Shin, B.H.; Lee, J.S.; Park, H.T.; Kim, P.; Lee, J.H.; Choi, Y. Saikosaponin D Ameliorates Mechanical Hypersensitivity in Animal Models of Neuropathic Pain. Planta Med. Int. Open 2020, 7, e145–e149.
- Li, Z.Y.; Jiang, Y.M.; Liu, Y.M.; Guo, Z.; Shen, S.N.; Liu, X.M.; Pan, R.L. Saikosaponin D acts against corticosterone-induced apoptosis via regulation of mitochondrial GR translocation and a GR-dependent pathway. Prog. Neuropsychopharmacol. Biol. Psychiatry 2014, 53, 80–89.
- Yuan, B.; Yang, R.; Ma, Y.; Zhou, S.; Zhang, X.; Liu, Y. A systematic review of the active saikosaponins and extracts isolated from Radix Bupleuri and their applications. Pharm. Biol. 2017, 55, 620–635.
- Lin, X.; Wu, S.; Wang, Q.; Shi, Y.; Liu, G.; Zhi, J.; Wang, F. Saikosaponin-D Reduces H2O2-Induced PC12 Cell Apoptosis by Removing ROS and Blocking MAPK-Dependent Oxidative Damage. Cell Mol. Neurobiol. 2016, 36, 1365–1375.
- Sun, T.; Song, W.G.; Fu, Z.J.; Liu, Z.H.; Liu, Y.M.; Yao, S.L. Alleviation of neuropathic pain by intrathecal injection of antisense oligonucleotides to p65 subunit of NF-κB. Br. J. Anaesth. 2006, 97, 553–558.
- Zhou, X.; Cheng, H.; Xu, D.; Yin, Q.; Cheng, L.; Wang, L.; Song, S.; Zhang, M. Attenuation of neuropathic pain by saikosaponin a in a rat model of chronic constriction injury. Neurochem. Res. 2014, 39, 2136–2142.
- Ramesh, G.; MacLean, A.G.; Philipp, M.T. Cytokines and chemokines at the crossroads of neuroinflammation, neurodegeneration, and neuropathic pain. Mediat. Inflamm. 2013, 2013, 480739.
- Lu, C.N.; Yuan, Z.G.; Zhang, X.L.; Yan, R.; Zhao, Y.Q.; Liao, M.; Chen, J.X. Saikosaponin a and its epimer saikosaponin d exhibit anti-inflammatory activity by suppressing activation of NF-kappaB signaling pathway. Int. Immunopharmacol. 2012, 14, 121–126.
- Zhu, J.; Luo, C.; Wang, P.; He, Q.; Zhou, J.; Peng, H. Saikosaponin A mediates the inflammatory response by inhibiting the MAPK and NF-kappaB pathways in LPS-stimulated RAW 264.7 cells. Exp. Ther. Med. 2013, 5, 1345–1350.
- Mao, X.; Miao, G.; Tao, X.; Hao, S.; Zhang, H.; Li, H.; Hou, Z.; Tian, R.; Lu, T.; Ma, J.; et al. Saikosaponin a protects TBI rats after controlled cortical impact and the underlying mechanism. Am. J. Transl. Res. 2016, 8, 133–141.
- Yoon, S.S.; Seo, J.W.; Ann, S.H.; Kim, H.Y.; Kim, H.S.; Cho, H.Y.; Yun, J.; Chung, E.Y.; Koo, J.S.; Yang, C.H. Effects of saikosaponin A on cocaine self-administration in rats. Neurosci. Lett. 2013, 555, 198–202.
- Yoon, S.S.; Kim, H.S.; Cho, H.Y.; Yun, J.; Chung, E.Y.; Jang, C.G.; Kim, K.J.; Yang, C.H. Effect of saikosaponin A on maintenance of intravenous morphine self-administration. Neurosci. Lett. 2012, 529, 97–101.
- Ahmadimoghaddam, D.; Zarei, M.; Mohammadi, S.; Izadidastenaei, Z.; Salehi, I. Bupleurum falcatum L. alleviates nociceptive and neuropathic pain: Potential mechanisms of action. J. Ethnopharmacol. 2021, 273, 113990.
- Minett, M.S.; Nassar, M.A.; Clark, A.K.; Passmore, G.; Dickenson, A.H.; Wang, F.; Malcangio, M.; Wood, J.N. Distinct Nav1.7-dependent pain sensations require different sets of sensory and sympathetic neurons. Nat. Commun. 2012, 3, 791.
- Xu, Y.; Yu, Y.; Wang, Q.; Li, W.; Zhang, S.; Liao, X.; Liu, Y.; Su, Y.; Zhao, M.; Zhang, J. Active components of Bupleurum chinense and Angelica biserrata showed analgesic effects in formalin induced pain by acting on Nav1.7. J. Ethnopharmacol. 2021, 269, 113736.
- Chen, Z.; Liu, L.; Gao, C.; Chen, W.; Vong, C.T.; Yao, P.; Yang, Y.; Li, X.; Tang, X.; Wang, S.; et al. Astragali Radix (Huangqi): A promising edible immunomodulatory herbal medicine. J. Ethnopharmacol. 2020, 258, 112895.
- Fu, J.; Wang, Z.; Huang, L.; Zheng, S.; Wang, D.; Chen, S.; Zhang, H.; Yang, S. Review of the botanical characteristics, phytochemistry, and pharmacology of Astragalus membranaceus (Huangqi). Phytother. Res. 2014, 28, 1275–1283.
- Song, J.Z.; Mo, S.F.; Yip, Y.K.; Qiao, C.F.; Han, Q.B.; Xu, H.X. Development of microwave assisted extraction for the simultaneous determination of isoflavonoids and saponins in radix astragali by high performance liquid chromatography. J. Sep. Sci. 2007, 30, 819–824.
- Ma, X.Q.; Shi, Q.; Duan, J.A.; Dong, T.T.; Tsim, K.W. Chemical analysis of Radix Astragali (Huangqi) in China: A comparison with its adulterants and seasonal variations. J. Agric. Food Chem. 2002, 50, 4861–4866.
- Wang, Y.; Liu, L.; Ma, Y.; Guo, L.; Sun, Y.; Liu, Q.; Liu, J. Chemical Discrimination of Astragalus mongholicus and Astragalus membranaceus Based on Metabolomics Using UHPLC-ESI-Q-TOF-MS/MS Approach. Molecules 2019, 24, 4064.
- Liu, D.-l.; Bao, H.-Y.; Liu, Y. Progress on Chemical Constituents and Pharmacological Effects of Astragali Radix in Recent Five Years. Food Drug 2014, 16, 68–70.
- Polat, E.; Bedir, E.; Perrone, A.; Piacente, S.; Alankus-Caliskan, O. Triterpenoid saponins from Astragalus wiedemannianus Fischer. Phytochemistry 2010, 71, 658–662.
- Verotta, L.; Guerrini, M.; El-Sebakhy, N.A.; Assad, A.M.; Toaima, S.M.; Radwan, M.M.; Luo, Y.D.; Pezzuto, J.M. Cycloartane and oleanane saponins from egyptian astragalus spp. as modulators of lymphocyte proliferation. Planta Med. 2002, 68, 986–994.
- Zhang, J.; Wu, C.; Gao, L.; Du, G.; Qin, X. Astragaloside IV derived from Astragalus membranaceus: A research review on the pharmacological effects. Adv. Pharm. 2020, 87, 89–112.
- Yang, F.; Xiao, X.; Lee, B.H.; Vu, S.; Yang, W.; Yarov-Yarovoy, V.; Zheng, J. The conformational wave in capsaicin activation of transient receptor potential vanilloid 1 ion channel. Nat. Commun. 2018, 9, 2879.
- Tominaga, M.; Caterina, M.J.; Malmberg, A.B.; Rosen, T.A.; Gilbert, H.; Skinner, K.; Raumann, B.E.; Basbaum, A.I.; Julius, D. The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron 1998, 21, 531–543.
- Mitchell, K.; Bates, B.D.; Keller, J.M.; Lopez, M.; Scholl, L.; Navarro, J.; Madian, N.; Haspel, G.; Nemenov, M.I.; Iadarola, M.J. Ablation of rat TRPV1-expressing Adelta/C-fibers with resiniferatoxin: Analysis of withdrawal behaviors, recovery of function and molecular correlates. Mol. Pain 2010, 6, 94.
- Li, X.; Kang, L.; Li, G.; Zeng, H.; Zhang, L.; Ling, X.; Dong, H.; Liang, S.; Chen, H. Intrathecal leptin inhibits expression of the P2X2/3 receptors and alleviates neuropathic pain induced by chronic constriction sciatic nerve injury. Mol. Pain 2013, 9, 65.
- Lin, J.; Li, G.; Den, X.; Xu, C.; Liu, S.; Gao, Y.; Liu, H.; Zhang, J.; Li, X.; Liang, S. VEGF and its receptor-2 involved in neuropathic pain transmission mediated by P2X2/3 receptor of primary sensory neurons. Brain Res. Bull. 2010, 83, 284–291.
- Shi, G.B.; Fan, R.; Zhang, W.; Yang, C.; Wang, Q.; Song, J.; Gao, Y.; Hou, M.X.; Chen, Y.F.; Wang, T.C.; et al. Antinociceptive activity of astragaloside IV in the animal model of chronic constriction injury. Behav. Pharm. 2015, 26, 436–446.
- Dong, Z.Q.; Ma, F.; Xie, H.; Wang, Y.Q.; Wu, G.C. Down-regulation of GFRalpha-1 expression by antisense oligodeoxynucleotide attenuates electroacupuncture analgesia on heat hyperalgesia in a rat model of neuropathic pain. Brain Res. Bull. 2006, 69, 30–36.
- Dong, Z.Q.; Ma, F.; Xie, H.; Wang, Y.Q.; Wu, G.C. Changes of expression of glial cell line-derived neurotrophic factor and its receptor in dorsal root ganglions and spinal dorsal horn during electroacupuncture treatment in neuropathic pain rats. Neurosci. Lett. 2005, 376, 143–148.
- Cheng, C.Y.; Yao, C.H.; Liu, B.S.; Liu, C.J.; Chen, G.W.; Chen, Y.S. The role of astragaloside in regeneration of the peripheral nerve system. J. Biomed. Mater. Res. A 2006, 76, 463–469.
- Zhang, X.; Chen, J. The mechanism of astragaloside IV promoting sciatic nerve regeneration. Neural Regen. Res. 2013, 8, 2256–2265.
- Shen, Y.; Meiri, K. GAP-43 dependency defines distinct effects of netrin-1 on cortical and spinal neurite outgrowth and directional guidance. Int. J. Dev. Neurosci. 2012, 31, 11–20.
- Mendonca, H.R.; Araujo, S.E.; Gomes, A.L.; Sholl-Franco, A.; da Cunha Faria Melibeu, A.; Serfaty, C.A.; Campello-Costa, P. Expression of GAP-43 during development and after monocular enucleation in the rat superior colliculus. Neurosci. Lett. 2010, 477, 23–27.
- Yu, J.; Zhang, Y.; Sun, S.; Shen, J.; Qiu, J.; Yin, X.; Yin, H.; Jiang, S. Inhibitory effects of astragaloside IV on diabetic peripheral neuropathy in rats. Can. J. Physiol. Pharm. 2006, 84, 579–587.
- Davidson, J.A. Treatment of the patient with diabetes: Importance of maintaining target HbA(1c) levels. Curr. Med. Res. Opin. 2004, 20, 1919–1927.
- Yi, T.; Fan, L.L.; Chen, H.L.; Zhu, G.Y.; Suen, H.M.; Tang, Y.N.; Zhu, L.; Chu, C.; Zhao, Z.Z.; Chen, H.B. Comparative analysis of diosgenin in Dioscorea species and related medicinal plants by UPLC-DAD-MS. BMC Biochem. 2014, 15, 19.
- Arya, P.; Kumar, P. Diosgenin a steroidal compound: An emerging way to cancer management. J. Food Biochem. 2021, 45, e14005.
- Chen, Y.; Tang, Y.-M.; Yu, S.-L.; Han, Y.-W.; Kou, J.-P.; Liu, B.-L.; Yu, B.-Y. Advances in the pharmacological activities and mechanisms of diosgenin. Chin. J. Nat. Med. 2015, 13, 578–587.
- Al-Habori, M.; Raman, A.; Lawrence, M.J.; Skett, P. In vitro effect of fenugreek extracts on intestinal sodium-dependent glucose uptake and hepatic glycogen phosphorylase A. Int. J. Exp. Diabetes Res. 2001, 2, 91–99.
- Fan, R.; He, W.; Fan, Y.; Xu, W.; Xu, W.; Yan, G.; Xu, S. Recent advances in chemical synthesis, biocatalysis, and biological evaluation of diosgenin derivatives—A review. Steroids 2022, 180, 108991.
- Fernandes, P.; Cruz, A.; Angelova, B.; Pinheiro, H.M.; Cabral, J.M.S. Microbial conversion of steroid compounds: Recent developments. Enzym. Microb. Tech. 2003, 32, 688–705.
- Obrosova, I.G. Update on the pathogenesis of diabetic neuropathy. Curr. Diabetes Rep. 2003, 3, 439–445.
- Kiasalari, Z.; Rahmani, T.; Mahmoudi, N.; Baluchnejadmojarad, T.; Roghani, M. Diosgenin ameliorates development of neuropathic pain in diabetic rats: Involvement of oxidative stress and inflammation. Biomed. Pharm. 2017, 86, 654–661.
- Kasznicki, J.; Kosmalski, M.; Sliwinska, A.; Mrowicka, M.; Stanczyk, M.; Majsterek, I.; Drzewoski, J. Evaluation of oxidative stress markers in pathogenesis of diabetic neuropathy. Mol. Biol. Rep. 2012, 39, 8669–8678.
- Vincent, A.M.; Russell, J.W.; Low, P.; Feldman, E.L. Oxidative stress in the pathogenesis of diabetic neuropathy. Endocr. Rev. 2004, 25, 612–628.
- Wang, C.; Ning, L.P.; Wang, Y.H.; Zhang, Y.; Ding, X.L.; Ge, H.Y.; Arendt-Nielsen, L.; Yue, S.W. Nuclear factor-kappa B mediates TRPV4-NO pathway involved in thermal hyperalgesia following chronic compression of the dorsal root ganglion in rats. Behav. Brain Res. 2011, 221, 19–24.
- Zhang, Y.P.; Song, C.Y.; Yuan, Y.; Eber, A.; Rodriguez, Y.; Levitt, R.C.; Takacs, P.; Yang, Z.; Goldberg, R.; Candiotti, K.A. Diabetic neuropathic pain development in type 2 diabetic mouse model and the prophylactic and therapeutic effects of coenzyme Q10. Neurobiol. Dis. 2013, 58, 169–178.
- Kumar, A.; Negi, G.; Sharma, S.S. Suppression of NF-κB and NF-κB regulated oxidative stress and neuroinflammation by BAY 11-7082 (IκB phosphorylation inhibitor) in experimental diabetic neuropathy. Biochimie 2012, 94, 1158–1165.
- Kawamura, N.; Dyck, P.J.; Schmeichel, A.M.; Engelstad, J.K.; Low, P.A.; Dyck, P.J. Inflammatory mediators in diabetic and non-diabetic lumbosacral radiculoplexus neuropathy. Acta Neuropathol. 2008, 115, 231–239.
- Gao, M.; Chen, L.; Yu, H.; Sun, Q.; Kou, J.; Yu, B. Diosgenin down-regulates NF-kappaB p65/p50 and p38MAPK pathways and attenuates acute lung injury induced by lipopolysaccharide in mice. Int. Immunopharmacol. 2013, 15, 240–245.
- Gao, Z.; Feng, Y.; Ju, H. The Different Dynamic Changes of Nerve Growth Factor in the Dorsal Horn and Dorsal Root Ganglion Leads to Hyperalgesia and Allodynia in Diabetic Neuropathic Pain. Pain Physician 2017, 20, E551–E561.
- Calissano, P.; Amadoro, G.; Matrone, C.; Ciafrè, S.; Marolda, R.; Corsetti, V.; Ciotti, M.T.; Mercanti, D.; Di Luzio, A.; Severini, C.; et al. Does the term ‘trophic’ actually mean anti-amyloidogenic? The case of NGF. Cell Death Differ. 2010, 17, 1126–1133.
- Kang, T.H.; Moon, E.; Hong, B.N.; Choi, S.Z.; Son, M.; Park, J.H.; Kim, S.Y. Diosgenin from Dioscorea nipponica ameliorates diabetic neuropathy by inducing nerve growth factor. Biol. Pharm. Bull. 2011, 34, 1493–1498.
- Zhao, W.X.; Wang, P.F.; Song, H.G.; Sun, N. Diosgenin attenuates neuropathic pain in a rat model of chronic constriction injury. Mol. Med. Rep. 2017, 16, 1559–1564.
- Lee, B.K.; Kim, C.J.; Shin, M.S.; Cho, Y.S. Diosgenin improves functional recovery from sciatic crushed nerve injury in rats. J. Exerc. Rehabil. 2018, 14, 566–572.
- Byun, Y.H.; Lee, M.H.; Kim, S.S.; Kim, H.; Chang, H.K.; Lee, T.H.; Jang, M.H.; Shin, M.C.; Shin, M.S.; Kim, C.J. Treadmill running promotes functional recovery and decreases brain-derived neurotrophic factor mRNA expression following sciatic crushed nerve injury in rats. J. Sports Med. Phys. Fit. 2005, 45, 222–228.
- Liu, M.; Luo, F.; Qing, Z.; Yang, H.; Liu, X.; Yang, Z.; Zeng, J. Chemical Composition and Bioactivity of Essential Oil of Ten Labiatae Species. Molecules 2020, 25, 4862.
- Palida, A.; Mi, R.; Cong, Y.; Yi, B.; Wang, X. Isolation and identification of chemical constituents in Ocimum bacilicum. West China J. Pharm. Sci. 2007, 22, 489–490.
- Kaur, G.; Bali, A.; Singh, N.; Jaggi, A.S. Ameliorative potential of Ocimum sanctum in chronic constriction injury-induced neuropathic pain in rats. An. Acad. Bras. Ciênc. 2015, 87, 417–429.
- Anandjiwala, S.; Kalola, J.; Rajani, M. Quantification of eugenol, luteolin, ursolic acid, and oleanolic acid in black (Krishna Tulasi) and green (Sri Tulasi) varieties of Ocimum sanctum Linn. using high-performance thin-layer chromatography. J. AOAC Int. 2006, 89, 1467–1474.
- Rao, A.R.; Veeresham, C.; Asres, K. In vitro and in vivo inhibitory activities of four Indian medicinal plant extracts and their major components on rat aldose reductase and generation of advanced glycation endproducts. Phytother. Res. 2013, 27, 753–760.
- Carrasco, C.; Naziroǧlu, M.; Rodríguez, A.B.; Pariente, J.A. Neuropathic Pain: Delving into the Oxidative Origin and the Possible Implication of Transient Receptor Potential Channels. Front. Physiol. 2018, 9, 95.
- Bourinet, E.; Altier, C.; Hildebrand, M.E.; Trang, T.; Salter, M.W.; Zamponi, G.W. Calcium-permeable ion channels in pain signaling. Physiol. Rev. 2014, 94, 81–140.
- Katsuyama, Y.; Sato, Y.; Okano, Y.; Masaki, H. Intracellular oxidative stress induced by calcium influx initiates the activation of phagocytosis in keratinocytes accumulating at S-phase of the cell cycle after UVB irradiation. J. Dermatol. Sci. 2021, 103, 41–48.
- Gibson, G.E. Interactions of oxidative stress with cellular calcium dynamics and glucose metabolism in Alzheimer’s disease. Free Radic. Biol. Med. 2002, 32, 1061–1070.
- Goodwin, J.; Nath, S.; Engelborghs, Y.; Pountney, D.L. Raised calcium and oxidative stress cooperatively promote alpha-synuclein aggregate formation. Neurochem. Int. 2013, 62, 703–711.
- Carbonera, D.; Azzone, G.F. Permeability of inner mitochondrial membrane and oxidative stress. Biochim. Biophys. Acta 1988, 943, 245–255.
- George, S.; Chaturvedi, P. Protective role of Ocimum canum plant extract in alcohol-induced oxidative stress in albino rats. Br. J. Biomed. Sci. 2008, 65, 80–85.
- Kelm, M.A.; Nair, M.G.; Strasburg, G.M.; DeWitt, D.L. Antioxidant and cyclooxygenase inhibitory phenolic compounds from Ocimum sanctum Linn. Phytomedicine 2000, 7, 7–13.
- Balanehru, S.; Nagarajan, B. Protective effect of oleanolic acid and ursolic acid against lipid peroxidation. Biochem. Int. 1991, 24, 981–990.
- Muthuraman, A.; Diwan, V.; Jaggi, A.S.; Singh, N.; Singh, D. Ameliorative effects of Ocimum sanctum in sciatic nerve transection-induced neuropathy in rats. J. Ethnopharmacol. 2008, 120, 56–62.
- Kaur, G.; Jaggi, A.S.; Singh, N. Exploring the potential effect of Ocimum sanctum in vincristine-induced neuropathic pain in rats. J. Brachial Plex. Peripher. Nerve Inj. 2010, 5, 3.
- Hu, S.; Wu, Y.; Zhao, B.; Hu, H.; Zhu, B.; Sun, Z.; Li, P.; Du, S. Panax notoginseng Saponins Protect Cerebral Microvascular Endothelial Cells against Oxygen-Glucose Deprivation/Reperfusion-Induced Barrier Dysfunction via Activation of PI3K/Akt/Nrf2 Antioxidant Signaling Pathway. Molecules 2018, 23, 2781.
- Neco, P.; Rose, B.; Huynh, N.; Zhang, R.; Bridge, J.H.; Philipson, K.D.; Goldhaber, J.I. Sodium-calcium exchange is essential for effective triggering of calcium release in mouse heart. Biophys. J. 2010, 99, 755–764.
- Sarkar, A.; Pandey, D.N.; Pant, M.C. A report on the effects of Ocimum sanctum (Tulsi) leaves and seeds on blood and urinary uric acid, urea and urine volume in normal albino rabbits. Indian J. Physiol. Pharmacol. 1990, 34, 61–62.
- Dalbeth, N.; Choi, H.K.; Joosten, L.A.B.; Khanna, P.P.; Matsuo, H.; Perez-Ruiz, F.; Stamp, L.K. Gout. Nat. Rev. Dis. Prim. 2019, 5, 69.
- Khanna, N.; Bhatia, J. Antinociceptive action of Ocimum sanctum (Tulsi) in mice: Possible mechanisms involved. J. Ethnopharmacol. 2003, 88, 293–296.
More