Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 3 by Jessie Wu and Version 2 by Jessie Wu.
T cell differentiation is a tightly regulated process. Recent studies have shown that epigenetics plays a significant role at all stages of the differentiation process. This entry briefly introduces the de different stages of T cell differentiation and will discussre introduced and recent findings on the epigenetic regulation of this process will be discussed. The epigenetic modifications associated with T cell differentiation related to cancer are discussed as well.
- epigenetics
- T cell differentiation
- cancer
- autoimmune disease
- Immunology
1. Generation of Differentiated T Cells
Mature T cells originate in the thymus and after going through various developmental stages they exit the thymus to circulate in the peripheral lymphoid organs including the spleen and lymph nodes . Originating in the bone marrow or fetal liver, thymic seeding progenitor cells (TSPs) (also known as early thymic progenitor cells (ETPs) or DN1 cells (CD4− CD8− double negative; DN)) enter the thymus, where they progress through four DN stages (DN1-4) (Figure 1) [1]. DN3 cells expressing the pre-TCR (pre-T cell receptor) transit to the DP (CD4+ CD8+, double positive; DP) stage (Figure 1) [1]. In the cortex, DP thymocytes undergo a process of positive and negative selection which is determined by TCR signaling in response to binding to self-peptide-self-major histocompatibility complexes (self-pMHC) and then commit to either the CD4+ or CD8+ SP (single positive; SP) lineage (Figure 1) [1]. Immature SP thymocytes undergo further differentiation to generate either MHC Class II restricted CD4 SP helper or MHC Class I restricted CD8 SP cytotoxic lineage cells [1].
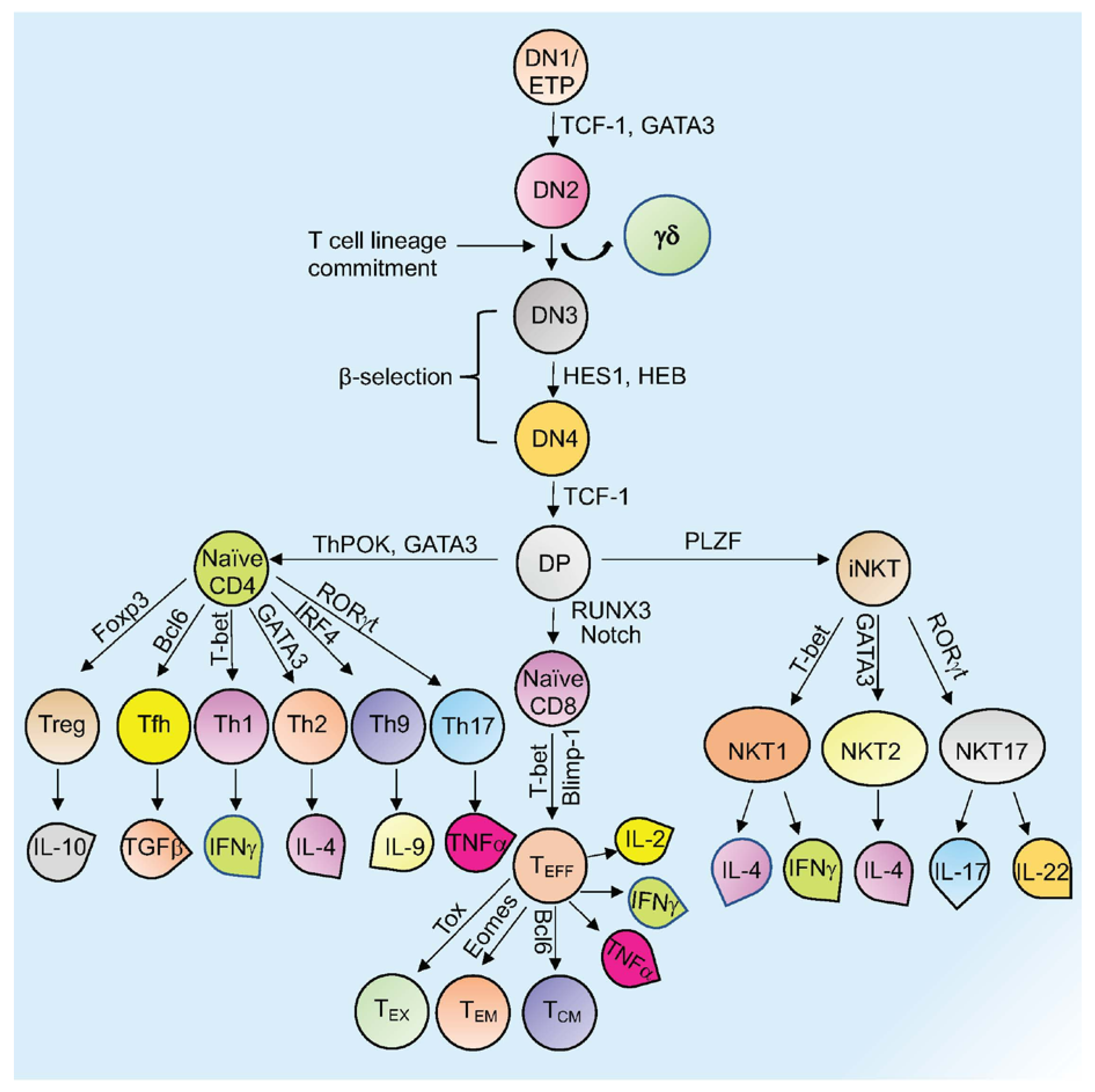
Figure 1. Overview of T cell development and differentiation. Schematic representation of thymopoiesis in mice. Following a series of DN (1–4) stages, DP cells develop into naïve CD4+, naïve CD8+, or natural killer T cells (NKT). Several transcription factors regulate this process. Different T cells secrete various cytokines to exert their activity. Signature transcription factors and cytokines designated to different cell types are shown. See the text for more details about each type of cell and their function (Tex= exhausted T cells; TEM = T effector memory T cells; TCM= T central memory T cells; TEFF = effector T cells; Th= T helper T cells; Tfh= T follicular helper T cells; Treg= regulatory T cells).
Other subsets of T cells generated in the thymus include regulatory T cells (Tregs) and natural killer T cells (NKT; Figure 1). Naïve human CD4+ T cells, which are now considered to be a heterogeneous population, can be subcategorized based on CD31 (also known as PECAM1) expression, into the youngest (recent thymus emigrant) naïve T cells (CD31+) and more established “mature” naïve T cells (CD31−) [2][3]. After encountering antigen, naïve CD4+ T cells differentiate into CD4+ effector T cells, which can take the form of well-defined subgroups that express distinct cytokine profiles: T helper 1 (Th-1), Th-2, Th-9, T follicular helper (Tfh) cells, and Th-17 cells (Figure 1) [4]. These differentiated subgroups are not necessarily definitive as some degree of plasticity can be observed, particularly if epigenetic control mechanisms are dysregulated (discussed below). Tregs, which comprise a separate CD4 lineage, work to prevent autoimmune diseases through suppressing the immune system’s responses to self-antigens [5]. These cells express the master regulator transcription factor (TFs) Foxp3. Tregs are subdivided based on their location and mode of generation, which includes thymus-derived Tregs (tTregs), naturally occurring Tregs (nTregs), induced Tregs (iTregs), and peripherally induced Tregs (pTregs) [6][7][8][9][10][11]. CD8+ T cells that encounter antigen differentiate into effector T cells (TEFF or Cytotoxic T cells; CTLs) that produce cytokines and cytotoxic enzymes (perforin, granzyme B) that eliminate pathogens or target cells (including pathogen-infected host cells and tumor cells) and can develop into memory T cells (Figure 1) [12]. Memory T cells (which are generated from activated CD4+ and CD8+ T cells) persevere following the generation of a primary immune response and are able to mount an enhanced secondary response to the same antigen [12]. Memory T cells can be subdivided into effector memory T cells (TEM) or central memory T cells (TCM) based on different surface markers (Figure 1) [12]. Another type of T cell with limited effector functions is known as the exhausted T cell (Tex). Both CD4+ and CD8+ T cells can differentiate into Tex cells when encountering chronic infections and cancer (Figure 1) [13][14].
2. Epigenetic Changes during T Cell Differentiation
Epigenetic modifications greatly influence the functional differentiation of T cell subsets, including lineage commitment to short-lived effectors, long-term memory T cells, T regulatory cells, and other specific T cell populations. The following sections focus on the cooperation between epigenetic changes and transcriptional programs related to different subsets of T cell differentiation. Major epigenetic regulators associated with T cell differentiation are summarized in Table 1.
Table 1. Major epigenetic regulators associated with T cell differentiation.
| Regulator | Cell Types | Modification/Function | References |
|---|---|---|---|
| Dot1L | Th-2, CD8+ | H3K79me2 | [12][13] |
| Menin | Th-2 | Menin/TrxG complex promotes H3K4me3 | [14][15][16][17][18] |
| Cxxc1 | Th-1, Th-2, Th-17 | Cxxc1/TrxG complex inhibits/promotes H3K4me3 in gene specific manner | [19][20][21][22] |
| ATF7ip | Th-17 | ATF7ip/SETDB1 complex promotes H3K9me3 | [23][24] |
| Utx | Th-17, iNKT | Histone demethylase | [25][26][27][28] |
| Hdac3 | CD4+, CD8+, iNKT | Histone deacetylation | [29][30][31] |
| Tcf1/ Lef1 | CD4+, CD8+ | Histone deacetylation | [32] |
| Gcn5 | Th-1, Th-17, iNKT | Histone acetylation | [33][34] |
| Rcor1 | Treg | Rcor1 is a part of CoREST complex which mediates histone deacetylation | [35][36] |
| Ezh2 | CD8+, iNKT | H3K27me3 | [13][26][37] |
| Lsd1 | CD8+ CTLs | H3K4 and H3K79 demethylase | [38][39][40] |
| Tox | Exhausted CD8+ | Binds to epigenetic regulators to change gene expression | [41][42][43] |
2.1. Epigenetic Control of CD4+ T Cell Differentiation
Naïve T cells manifest a unique epigenetic landscape [2]. One recent study in mice found that RTEs (recent thymic emigrants) and more “mature” naïve T cells exhibit different DNA methylation patterns at key cytokine (Il2 and Il4) loci, contributing to post- thymic naïve T cell maturation [44]. However, additional studies are required to fully understand the epigenetic mechanisms that regulate naïve T cell formation/maintenance and naïve T cell “maturation”. One report showed that CD4+ T helper (Th) cell differentiation is regulated by lysine methyltransferase (KMT) Dot1l-dependent di-methylation of lysine 79 of histone H3 (H3K79me2), which is associated with lineage-specific gene expression [12]. Loss of Dot1l (mediated by Cd4-Cre, which becomes active in thymocytes at the DP stage) leads to increased expression of Th-1-specific genes and overproduction of IFN-γ at the expense of Th-2 cell development, suggesting a central role for Dot1l in Th-2 cell lineage commitment and stability [12].Using Cd4-cre-driven conditional knockout (KO) mice, another recent report showed a contribution of Menin, a major component of the Trithorax group (TrxG) complex, in the acquisition and maintenance of Th-2 cell identity [14]. The TrxG complex catalyzes the trimethylation of H3K4, resulting in induction or maintenance of gene transcription [15]. It has been shown that Menin is an extremely specific partner for mixed lineage leukemia (MLL)1/2-containing H3K4 methyltransferase complexes [16] and that binding of the Menin/TrxG complex is required for the maintenance of Gata3 expression and Th-2 cytokine production in established Th-2 cells both in mice and humans [17][18]. Previous reports have demonstrated that Gata3 is critical for commitment of DP thymocytes to the CD4 SP lineage as well as CD4 Th-2 lineage generation [45][46]. To promote the differentiation of Th-2 cells, Gata3 directly binds to the promoters of the Il3 and Il5 genes, which allows for Th-2 cytokines to be expressed [47]. This research found that Menin supports memory Th-2 (mTh-2) (a subset of Th-2 cells that produce large amounts of IL-4 and IL-13 in response to antigenic re-stimulation) cell function and deletion of Menin results in a significant reduction in the number of mTh-2 cells compared with WT mice [14]. Mechanistically, they showed that Menin deficiency leads to a decrease in Gata3 expression due to reduced levels of H3K9ac and H3K4me3 at the upstream regions of the Gata3 proximal promoter [14]. Another recent report showed that Cxxc1, which can also be a subunit of the Trithorax complex, regulates the generation of functional Th-1/Th-2 cells since Cxxc1 epigenetically represses transcription of genes, including Trib3 and Klf2, that are required for these differentiation processes via binding to their promoter regions [19]. The authors showed that Cxxc1 depletion by tamoxifen inducible ER-Cre resulted in the hyperactivation of master transcription factors including T-bet in Th-1 cells and Gata3 in Th-2 cells and hyperproduction of Th-1 and Th-2 cytokines. They also showed that Cxxc1 indirectly controls the expression of Gata3 through direct binding and regulation of Klf2, which negatively regulates Gata3 expression [19]. By performing ChIP sequencing, they found that Cxxc1 directly binds to the Klf2 promoter region and that depletion of Cxxc1 resulted in a decrease in H3K4me3 levels at the Klf2 promoter [19]. These data suggest that although part of the Trithorax complex, different components have distinct functions in the differentiation and regulation of CD4+ Th-1 and Th-2 cells. Interestingly, another study using dLck-Cre showed that T cell-specific ablation of Cxxc1 results in the generation of severely defective Th-17 cells [20]. Mechanistically, they found that depletion of Cxxc1 results in decreased IL-6Rα expression since Cxxc1 maintains H3K4me3 modification at its promoter [20]. Depletion of IL-6Rα disrupts IL-6/STAT3 signaling, which is known to initiate Th-17 cell differentiation, resulting in the production of Treg cells instead of Th-17 cells, suggesting that Cxxc1 keeps a balance between these two lineages [20][21][22]. The contradiction between the two reports, i.e., Th-1 and Th-2 production [19] vs. Th-17 cells [20] generation due to Cxxc1 ablation, might be due to the Cre system (ER-Cre vs. dLck-Cre) that was used as the former targets mature T cells whereas the latter acts on immature pre-selection thymocytes. Another study tested the effects of a new BET (bromodomain and extra-terminal) inhibitor, OTX015, on CD4+ Th-17 cells [48]. BET proteins are epigenetic regulators that recognize and bind to acetylated histones in chromatin and that regulate gene expression. The authors showed that OTX015 has anti-inflammatory effects via suppression of CD4+ T cell proliferation in both mice and humans and suppresses the production of Th-17 cytokines (IL-17 in particular) in humans [48]. These data suggest that BET plays an important role in CD4+ Th-17 cell generation and persistence. Another report identified ATF7ip (activating transcription factor 7 interacting protein, also known as MCAF1 or mAM), an epigenetic regulator responsible for repressive H3K9me3 marks via its histone methyltransferase-binding partners SETDB1/ESET [23], as a crucial regulator of Th-17 differentiation [24]. Depletion of Atf7ip (Cd4-Cre) resulted in impaired Th-17 differentiation and enhanced production of IL-2 in response to T cell receptor (TCR) stimulation [24]. They further showed that ATF7ip acts as an IL-2 inhibitor via suppression of Il2 gene expression through H3K9me3 deposition in the Il2-Il21 intergenic region [24]. Future studies will determine if ATF7ip inhibition could be useful for the treatment of Th-17-mediated autoimmune diseases. Using a combined chemico-genetic approach, another recent study showed that the histone H3K27 demethylases KDM6A (UTX) and KDM6B (JMJD3) function as central regulators of human Th subsets [25]. The authors utilized the pan-KDM6 inhibitor GSK-J4, which increases H3K27me3 decoration genome wide and suppresses the expression of RORγt during Th-17 differentiation in human CD4+ T cells [25]. They showed that KDM6 inhibition in mature Th-17 cells leads to reduced mitochondrial biogenesis, resulting in metabolic reprogramming and reduced expression of key metabolic TFs, such as PPRC1, which ultimately showed anti-inflammatory effects [25].
Another study also showed the importance of epigenetic control of metabolism in follicular helper T (Tfh) cell differentiation [49]. This report showed that the E3 ubiquitin ligase, Von Hippel–Lindau (VHL), was indispensable for Tfh cell development and function [49]. Using VHL conditional knockout mice (Cd4-Cre) and acute virus infection or antigen immunization, the authors documented that VHL positively regulates Tfh cell development and function from the very initiation stages [49]. Mechanistically, they found that VHL acts through the HIF-1α (hypoxia-inducible factor 1α)-dependent glycolysis pathway to promote the development of Tfh cells. VHL depletion leads to enhanced glycolytic activity via GAPDH, which reduces ICOS expression, a critical molecule for Tfh development, through epigenetic regulation of N6-methyladenosine (m6A) in Tfh cells [49]. Thus, this research points out the interplay between metabolism and epigenetic control on T cell differentiation [49].
Other than DNA demethylation or histone lysine methylation, histone acetylation/deacetylation also plays a major role in CD4+ cell fate determination [29]. Using Cd4-cre-mediated Hdac3 conditional knockout in mice, one study showed that compared to wild-type mice, the peripheral numbers of CD4+ and CD8+ T cells are significantly reduced as they enter the long-lived naïve T cell pool, suggesting a block at the RTE (recent thymic emigrant) stage of T cell maturation [29]. A previous study showed that Cd4-Cre-mediated Hdac3 conditional knockout mice have normal development of conventional T cells but exhibit a block in iNKT cell development [30]. Here, the authors further showed that Hdac3-deficient naïve peripheral T cells (both CD4+ and CD8+) have a defect in functional maturation, since these cells fail to produce TNF upon TCR/CD28 stimulation, suggesting that Hdac3 is critical for functional T cell maturation [29].
Interestingly, another study showed that the transcription factors Tcf-1 (Tcf7) and Lef-1 have intrinsic HDAC activity and are essential for repressing CD4+ lineage-associated genes, including Cd4, Foxp3, and Rorc, in CD8+ T cells via histone deacetylation [32]. Tcf1- and Lef1-deficient CD8+ T cells have increased H3K27Ac and H3K9Ac marks due to the diminished intrinsic HDAC activity of Tcf-1- and Lef-1 and Tcf-1- and Lef-1-deficient CD8+ T cells lose the ability to suppress CD4+ lineage-specific genes [32]. They also performed homology modeling to predict the Tcf-1 domain that would match known HDACs. They found that the Tcf-1 HDAC domain is similar in structure to a region in the Hdac8 catalytic pocket [32]. Solving the structure of Tcf-1 and Lef-1 will help understand the molecular basis of their HDAC activity. One study using histone acetyltransferase Gcn5 (encoded by Kat2a) conditionally deleted mice (Lck-Cre) revealed that Gcn5 plays pivotal roles in multiple stages of T cell development and differentiation [33]. They found a developmental block at the DN3 thymocyte stage upon loss of Gcn5 expression [33]. The authors mainly focused on T cell activation and differentiation; therefore, further studies are needed to address the mechanistic role of Gcn5 in β-selection and T cell development. However, they showed that in vitro deletion of Kat2a by tamoxifen treatment of naïve CD4+ T cells (using ESR-Cre) resulted in a severe defect in CD4+ T cell proliferation [33]. Depletion of Gcn5 also resulted in impaired IL-2 production and perturbed differentiation of Th-1/Th-17 cells but not Th-2 and Treg cell differentiation [33]. Mechanistically, they found that Gcn5 acetylates histone H3K9 to promote IL-2 production and suggested that Gcn5 may be a target for the treatment of autoimmune diseases [33].
2.2. Epigenetic Regulation of CD8+ T Cell Differentiation
CD8+ T cells play a critical role in removing or killing intracellular pathogen-infected cells and tumor cells. A recent study using bacterial infection as a model, characterized the epigenetic landscapes of naïve, effector, and memory CD8+ T cells and identified TFs that promote CD8+ T cell differentiation [50]. They found that the TF YY1 (yin and yang-1) promotes adoption of an effector T cell phenotype while Nr3c1 (nuclear receptor subfamily 3 group C member 1; a glucocorticoid receptor) promotes a memory-progenitor cell phenotype [50]. Age plays an important role in determining the fate of naïve T cells. One recent study showed CD8+ T cells from older adults have significantly reduced accessibility to genes responsible for maintaining cellular quiescence [51]. The authors further showed that compared to young adults, older adult CD8+ T cells have reduced expression of the TFs YY1 and NRF1 (nuclear respiratory factor 1) due to less accessibility to their chromatin binding sites [51].
The role of Ezh2, which is the catalytic subunit of the PRC2 complex that mediates di- and trimethylation of H3K27, is well established for CD8+ T cell differentiation and function and is reviewed elsewhere [52][53]. A new study showed that Ezh2 is required for differentiation of terminal effector CD8+ T cells but not for memory CD8+ T cell formation, although Ezh2-deficient CD8+ memory T cells are unable to clear infection, suggesting it is required for protective immunity [37]. Further study is needed to identify the underlying molecular mechanism behind this phenotype. As compared to the PRC2 complex, which creates repressive marks, Dot1l is associated with the positive regulation of gene transcription via mono-, di-, and trimethylation of histone H3K79 [13]. Using Lck-Cre-mediated conditional Dot1L knockout mice, the authors showed that loss of H3K79me2 in T cells leads to loss of naïve CD8+ T cells due to premature differentiation toward a memory-like state independent of antigen exposure [13]. Mechanistically, they showed Ezh2 is a target of Dot1l and loss of Dot1l affects Ezh2 function, leading to de-repression of a subset of PRC2 targets, which are actively repressed in control mice. They also showed that Dot1l ablation leads to repression of developmentally regulated genes, but the exact mechanism is yet to be determined [13]. Another study showed that CD40 ligand (CD40l), which is a member of the TNF superfamily proteins and helps in producing and maintaining cytotoxic T cells [54], is epigenetically suppressed in CD8+ cytotoxic T cells [55]. The promoter region of the Cd40lg gene was modified by methylation of CpG dinucleotides and suppressive histone lysine methylation marks. Mechanistically, they showed that enforced expression of the TF Thpok (Zbtb7b) leads to increased CD40l expression in CD8+ cytotoxic T cells via inhibition of Cxxc5, which interacts with the histone-lysine N-methyltransferase SUV39H1 to induce H3K9 methylation [55].
One recent report demonstrated that conditional deletion of Hdac3 in CD8+ T cells (using CD8 lineage-specific E8I-Cre) leads to more cytotoxicity while total numbers of CD8+ T cells were unaffected, suggesting that Hdac3 acts as a negative regulator for CD8+ T cell cytotoxic activity [31]. They also found that Hdac3 is required for activation of CD8+ T cells following an acute LCMV (lymphocytic choriomeningitis virus) infection [31]. Mechanistically, they found that Hdac3 depletion leads to an increase in enrichment of H3K27ac decoration at the Runx3 and Gzmb (granzyme B) genomic loci, which was essential for CD8+ T cell cytotoxicity [31], whereas increased H3K27ac at Prdm1 (Blimp-1), which is a transcriptional repressor that enhances terminal differentiation of effector CD8+ T cells during viral infection [56], was necessary for T cell persistence during activation [31].
3. Epigenetic Modification and Cancer
Several studies have shown that aberrant epigenetic modifications of T cells are associated with different cancers. LSD1 demethylates H3K4 and H3K79 and its expression is upregulated in many cancers including T cell acute lymphoblastic leukemia (T-ALL), breast, prostate, hepatocellular, and several others [57][58]. LSD1 inhibitors are currently under clinical trials for cancer treatment [59]. Recently, one study showed that in the case of triple-negative breast cancer (TNBC), administration of LSD1 inhibitor increases CD8+ cytotoxic T cell trafficking towards tumor sites with increased chemokine expression including CCL5, CXCL9, and CXCL10, which are known to recruit CD8+ T cells to tumors (Table 1) [38][60]. They further showed that LSD1 inhibitor treatment increases the active histone mark, H3K4me2, in the promoter regions of these chemokine genes, leading to more CD8+ T cell recruitment and disease remission [38]. Another study using a mouse breast cancer model showed that inhibition of nuclear Lsd1 (nLsd1) promotes infiltration of IFN-γ/TNF-α-expressing CD8+ T cells into tumors [39]. They also found, in dysfunctional “exhausted” CD8+ T cells, that the transcription factor Eomes and nLsd1 are co-expressed and that Lsd1 promotes T cell exhaustion by bivalent post-translational modification at lysine 641 of Eomes to prevent its nuclear entry [39]. Therefore, targeting nLSD1 may prevent T cell exhaustion and yield better therapeutic potential against tumors. Similar anti-tumor effects were obtained from a mouse melanoma study by using Lsd1 inhibitor or deleting Lsd1 (CRISPR/Cas9-mediated gene deletion) [40]. They found that Lsd1 ablation leads to activation of type 1 interferons (IFNs) and significant increases in both CD4+ and CD8+ T cells in melanoma tumors [40]. Mechanistically, they showed that ablation of Lsd1 leads to upregulation of ERV (endogenous retroviral element) transcription, resulting in activation of type-1 IFNs [40]. Taken together, these results clearly demonstrate that LSD1 plays an important role in effector T cell function, but its role in normal T cell development and differentiation is yet to be determined. One report showed that treatment with the DNA hypomethylating agent (HMA), decitabine (DAC), leads to increased CD8+ T cell tumor infiltration and inhibits tumor growth via CD8+ T cell-dependent activity in different mouse tumor models [61]. Using healthy human donors, the authors further showed that DAC treatment leads to T cell activation and expansion of the granzyme Bhigh, perforinhigh effector subpopulation and also increased expression of NFATc1/A, both of which contribute to the cytolytic activity of CD8+ T cells [61]. Collectively, this research identified that DNA methylation suppresses a subset of genes required for CD8+ T cell activation and that HMA treatment reprograms these genes to become activated, promoting cytolytic activity. Exhausted CD8+ T cells (Tex) have limited function as effector cells against chronic infections and cancer due to extensive transcriptional changes and increased co-expression of inhibitory receptors [62][63]. Recently, one study reported that deletion of the HMG-box transcription factor Tox (Cd4-Cre) leads to complete ablation of Tex formation (Table 1) [41]. Mechanistically, they showed that depletion of Tox leads to increased chromatin accessibility at genes linked to terminal CD8+ T effector cell differentiation, including Klrg1, Zeb2, Gzma, Gzmb, etc. They also showed that Tox directly binds with the acetyl transferase Kat7 as well as repressive epigenetic regulators (e.g., Dnmt1, Leo1), suggesting that Tox interacts with proteins involved in both the opening and closing of chromatin, leading to gene regulation [41]. Similar observations were made by another study that found that Tox and Tox2 cooperate with Nr4a (Nuclear Receptor Subfamily 4 Group A Member 1) transcription factors to promote CD8+ T cell exhaustion [42]. Finally, another group identified Tox as a critical modulator for the differentiation of tumor-specific T (TST) cells in a mouse model of liver cancer [43]. TST is a distinct state compared to functional effector or memory T cell states [64]. TSTs, which are mainly found in solid tumors, are dysfunctional as tumors progress despite their presence [65]. The authors showed that, in dysfunctional TST cells from tumors and in exhausted T cells obtained from chronic viral infection, Tox levels are significantly elevated [43]. Ablation of Tox (Lck-Cre) in TST cells in tumors diminished T cell exhaustion via downregulation of inhibitory receptor genes including Pdcd1, Tigit, Cd244, Entpd1, and Havcr2 through inaccessible chromatin [43]. Although Tox deletion abrogates T cell exhaustion, TST cells still remained dysfunctional, suggesting additional molecular mechanisms for exhaustion still remain to be identified [43]. Collectively, these studies shed light on CD8+ T cell exhaustion and establish the importance of Tox-mediated epigenetic modifications in this process. These findings also suggest that Tox may be a therapeutic target to decrease exhaustion of CD8+ T effector cells.4. Perspective and Future Directions
Recent studies have shed light on the importance of epigenetic regulation in the T cell differentiation process. Several mutations in epigenetic modifiers have been reported, which contribute to disease pathogenesis due to abnormal changes in gene expression required for T cell differentiation [66]. Specific inhibitors of chromatin remodeling proteins are currently under clinical trials, which may have novel treatment options for patients. It is also important to note that, as discussed in this review, some inhibitors are associated with unwanted toxicity or risk of autoimmune diseases. At the same time, various studies point out the rate-limiting factors that activate or suppress CTL functions. These findings may help identify novel approaches to improve immunotherapy for various cancers. Therefore, continuous efforts to understand the T cell differentiation process and the epigenetic regulators of this process will yield valuable information, which may be used for translational purposes.
References
- Dutta, A.; Zhao, B.; Love, P.E. New insights into TCR β-selection. Trends Immunol. 2021, 42, 735–750.
- Van den Broek, T.; Borghans, J.A.M.; van Wijk, F. The full spectrum of human naive T cells. Nat. Rev. Immunol. 2018, 18, 363–373.
- Silva, S.L.; Sousa, A.E. Establishment and Maintenance of the Human Naïve CD4+ T-Cell Compartment. Front. Pediatrics 2016, 4, 119.
- Zhu, J.; Yamane, H.; Paul, W.E. Differentiation of effector CD4 T cell populations. Annu. Rev. Immunol. 2010, 28, 445–489.
- Kondĕlková, K.; Vokurková, D.; Krejsek, J.; Borská, L.; Fiala, Z.; Ctirad, A. Regulatory T cells (TREG) and their roles in immune system with respect to immunopathological disorders. Acta Med. 2010, 53, 73–77.
- Fontenot, J.D.; Gavin, M.A.; Rudensky, A.Y. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 2003, 4, 330–336.
- Kitagawa, Y.; Ohkura, N.; Sakaguchi, S. Epigenetic control of thymic Treg-cell development. Eur. J. Immunol. 2015, 45, 11–16.
- Ono, M.; Yaguchi, H.; Ohkura, N.; Kitabayashi, I.; Nagamura, Y.; Nomura, T.; Miyachi, Y.; Tsukada, T.; Sakaguchi, S. Foxp3 controls regulatory T-cell function by interacting with AML1/Runx1. Nature 2007, 446, 685–689.
- Rudra, D.; deRoos, P.; Chaudhry, A.; Niec, R.E.; Arvey, A.; Samstein, R.M.; Leslie, C.; Shaffer, S.A.; Goodlett, D.R.; Rudensky, A.Y. Transcription factor Foxp3 and its protein partners form a complex regulatory network. Nat. Immunol. 2012, 13, 1010–1019.
- Iizuka-Koga, M.; Nakatsukasa, H.; Ito, M.; Akanuma, T.; Lu, Q.; Yoshimura, A. Induction and maintenance of regulatory T cells by transcription factors and epigenetic modifications. J. Autoimmun. 2017, 83, 113–121.
- Benoist, C.; Mathis, D. Treg cells, life history, and diversity. Cold Spring Harb. Perspect. Biol. 2012, 4, a007021.
- Scheer, S.; Runting, J.; Bramhall, M.; Russ, B.; Zaini, A.; Ellemor, J.; Rodrigues, G.; Ng, J.; Zaph, C. The Methyltransferase DOT1L Controls Activation and Lineage Integrity in CD4+ T Cells during Infection and Inflammation. Cell Rep. 2020, 33, 108505.
- Kwesi-Maliepaard, E.M.; Aslam, M.A.; Alemdehy, M.F.; Van Den Brand, T.; McLean, C.; Vlaming, H.; Van Welsem, T.; Korthout, T.; Lancini, C.; Hendriks, S.; et al. The histone methyltransferase DOT1L prevents antigen-independent differentiation and safeguards epigenetic identity of CD8+ T cells. Proc. Natl. Acad. Sci. USA 2020, 117, 20706–20716.
- Onodera, A.; Kiuchi, M.; Kokubo, K.; Kato, M.; Ogino, T.; Horiuchi, S.; Kanai, U.; Hirahara, K.; Nakayama, T. Menin Controls the Memory Th2 Cell Function by Maintaining the Epigenetic Integrity of Th2 Cells. J. Immunol. 2017, 199, 1153–1162.
- Schuettengruber, B.; Martinez, A.M.; Iovino, N.; Cavalli, G. Trithorax group proteins: Switching genes on and keeping them active. Nat. Rev. Mol. Cell Biol. 2011, 12, 799–814.
- Matkar, S.; Thiel, A.; Hua, X. Menin: A scaffold protein that controls gene expression and cell signaling. Trends Biochem. Sci. 2013, 38, 394–402.
- Onodera, A.; Yamashita, M.; Endo, Y.; Kuwahara, M.; Tofukuji, S.; Hosokawa, H.; Kanai, A.; Suzuki, Y.; Nakayama, T. STAT6-mediated displacement of polycomb by trithorax complex establishes long-term maintenance of GATA3 expression in T helper type 2 cells. J. Exp. Med. 2010, 207, 2493–2506.
- Nakata, Y.; Brignier, A.C.; Jin, S.; Shen, Y.; Rudnick, S.I.; Sugita, M.; Gewirtz, A.M. c-Myb, Menin, GATA-3, and MLL form a dynamic transcription complex that plays a pivotal role in human T helper type 2 cell development. Blood 2010, 116, 1280–1290.
- Kiuchi, M.; Onodera, A.; Kokubo, K.; Ichikawa, T.; Morimoto, Y.; Kawakami, E.; Takayama, N.; Eto, K.; Koseki, H.; Hirahara, K.; et al. The Cxxc1 subunit of the Trithorax complex directs epigenetic licensing of CD4+ T cell differentiation. J. Exp. Med. 2021, 218, e20201690.
- Lin, F.; Meng, X.; Guo, Y.; Cao, W.; Liu, W.; Xia, Q.; Hui, Z.; Chen, J.; Hong, S.; Zhang, X.; et al. Epigenetic initiation of the TH17 differentiation program is promoted by Cxxc finger protein 1. Sci. Adv. 2019, 5, eaax1608.
- Wei, L.; Laurence, A.; Elias, K.M.; O’Shea, J.J. IL-21 is produced by Th17 cells and drives IL-17 production in a STAT3-dependent manner. J. Biol. Chem. 2007, 282, 34605–34610.
- Camporeale, A.; Poli, V. IL-6, IL-17 and STAT3: A holy trinity in auto-immunity? Front. Biosci. (Landmark Ed.) 2012, 17, 2306–2326.
- Timms, R.T.; Tchasovnikarova, I.A.; Antrobus, R.; Dougan, G.; Lehner, P.J. ATF7IP-Mediated Stabilization of the Histone Methyltransferase SETDB1 Is Essential for Heterochromatin Formation by the HUSH Complex. Cell Rep. 2016, 17, 653–659.
- Sin, J.H.; Zuckerman, C.; Cortez, J.T.; Eckalbar, W.L.; Erle, D.J.; Anderson, M.S.; Waterfield, M.R. The epigenetic regulator ATF7ip inhibits Il2 expression, regulating Th17 responses. J. Exp. Med. 2019, 216, 2024–2037.
- Cribbs, A.P.; Terlecki-Zaniewicz, S.; Philpott, M.; Baardman, J.; Ahern, D.; Lindow, M.; Obad, S.; Oerum, H.; Sampey, B.; Mander, P.K.; et al. Histone H3K27me3 demethylases regulate human Th17 cell development and effector functions by impacting on metabolism. Proc. Natl. Acad. Sci. USA 2020, 117, 6056–6066.
- Dobenecker, M.-W.; Kim, J.K.; Marcello, J.; Fang, T.C.; Prinjha, R.; Bosselut, R.; Tarakhovsky, A. Coupling of T cell receptor specificity to natural killer T cell development by bivalent histone H3 methylation. J. Exp. Med. 2015, 212, 297–306.
- Beyaz, S.; Kim, J.H.; Pinello, L.; Xifaras, M.E.; Hu, Y.; Huang, J.; Kerenyi, M.A.; Das, P.P.; Barnitz, R.A.; Herault, A.; et al. The histone demethylase UTX regulates the lineage-specific epigenetic program of invariant natural killer T cells. Nat. Immunol. 2017, 18, 184–195.
- Meng, M.; Liu, H.; Chen, S.; Zhao, H.; Gao, X.; Zhang, J.; Chen, D. Methylation of H3K27 and H3K4 in key gene promoter regions of thymus in RA mice is involved in the abnormal development and differentiation of iNKT cells. Immunogenetics 2019, 71, 489–499.
- Hsu, F.-C.; Belmonte, P.J.; Constans, M.M.; Chen, M.W.; McWilliams, D.C.; Hiebert, S.W.; Shapiro, V.S. Histone Deacetylase 3 Is Required for T Cell Maturation. J. Immunol. 2015, 195, 1578–1590.
- Thapa, P.; Das, J.; McWilliams, D.; Shapiro, M.; Sundsbak, R.; Nelson-Holte, M.; Tangen, S.; Anderson, J.; Desiderio, S.; Hiebert, S.; et al. The transcriptional repressor NKAP is required for the development of iNKT cells. Nat. Commun. 2013, 4, 1582.
- Tay, R.E.; Olawoyin, O.; Cejas, P.; Xie, Y.; Meyer, C.A.; Ito, Y.; Weng, Q.Y.; Fisher, D.E.; Long, H.W.; Brown, M.; et al. Hdac3 is an epigenetic inhibitor of the cytotoxicity program in CD8 T cells. J. Exp. Med. 2020, 217, e20191453.
- Xing, S.; Li, F.; Zeng, Z.; Zhao, Y.; Yu, S.; Shan, Q.; Li, Y.; Phillips, F.C.; Maina, P.K.; Qi, H.H.; et al. Tcf1 and Lef1 transcription factors establish CD8+ T cell identity through intrinsic HDAC activity. Nat. Immunol. 2016, 17, 695–703.
- Gao, B.; Kong, Q.; Zhang, Y.; Yun, C.; Dent, S.Y.R.; Song, J.; Zhang, D.D.; Wang, Y.; Li, X.; Fang, D. The Histone Acetyltransferase Gcn5 Positively Regulates T Cell Activation. J. Immunol. 2017, 198, 3927–3938.
- Wang, Y.; Yun, C.; Gao, B.; Xu, Y.; Zhang, Y.; Wang, Y.; Kong, Q.; Zhao, F.; Wang, C.-R.; Dent, S.Y.R.; et al. The Lysine Acetyltransferase GCN5 Is Required for iNKT Cell Development through EGR2 Acetylation. Cell Rep. 2017, 20, 600–612.
- Xiong, Y.; Wang, L.; Di Giorgio, E.; Akimova, T.; Beier, U.H.; Han, R.; Trevisanut, M.; Kalin, J.H.; Cole, P.A.; Hancock, W.W. Inhibiting the coregulator CoREST impairs Foxp3+ Treg function and promotes antitumor immunity. J. Clin. Investig. 2020, 130, 1830–1842.
- Kalin, J.H.; Wu, M.; Gomez, A.V.; Song, Y.; Das, J.; Hayward, D.; Adejola, N.; Wu, M.; Panova, I.; Chung, H.J.; et al. Targeting the CoREST complex with dual histone deacetylase and demethylase inhibitors. Nat. Commun. 2018, 9, 53.
- Gray, S.M.; Amezquita, R.A.; Guan, T.; Kleinstein, S.H.; Kaech, S.M. Polycomb Repressive Complex 2-Mediated Chromatin Repression Guides Effector CD8+ T Cell Terminal Differentiation and Loss of Multipotency. Immunity 2017, 46, 596–608.
- Qin, Y.; Vasilatos, S.N.; Chen, L.; Wu, H.; Cao, Z.; Fu, Y.; Huang, M.; Vlad, A.M.; Lu, B.; Oesterreich, S.; et al. Inhibition of histone lysine-specific demethylase 1 elicits breast tumor immunity and enhances antitumor efficacy of immune checkpoint blockade. Oncogene 2019, 38, 390–405.
- Tu, W.J.; McCuaig, R.D.; Tan, A.H.Y.; Hardy, K.; Seddiki, N.; Ali, S.; Dahlstrom, J.E.; Bean, E.G.; Dunn, J.; Forwood, J.; et al. Targeting Nuclear LSD1 to Reprogram Cancer Cells and Reinvigorate Exhausted T Cells via a Novel LSD1-EOMES Switch. Front. Immunol. 2020, 11, 1228.
- Sheng, W.; Lafleur, M.W.; Nguyen, T.H.; Chen, S.; Chakravarthy, A.; Conway, J.R.; Li, Y.; Chen, H.; Yang, H.; Hsu, P.-H.; et al. LSD1 Ablation Stimulates Anti-tumor Immunity and Enables Checkpoint Blockade. Cell 2018, 174, 549–563.e19.
- Khan, O.; Giles, J.R.; McDonald, S.; Manne, S.; Ngiow, S.F.; Patel, K.P.; Werner, M.T.; Huang, A.C.; Alexander, K.A.; Wu, J.E.; et al. TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature 2019, 571, 211–218.
- Seo, H.; Chen, J.; González-Avalos, E.; Samaniego-Castruita, D.; Das, A.; Wang, Y.H.; López-Moyado, I.F.; Georges, R.O.; Zhang, W.; Onodera, A.; et al. TOX and TOX2 transcription factors cooperate with NR4A transcription factors to impose CD8+ T cell exhaustion. Proc. Natl. Acad. Sci. USA 2019, 116, 12410–12415.
- Scott, A.C.; Dündar, F.; Zumbo, P.; Chandran, S.S.; Klebanoff, C.A.; Shakiba, M.; Trivedi, P.; Menocal, L.; Appleby, H.; Camara, S.; et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature 2019, 571, 270–274.
- Berkley, A.M.; Hendricks, D.W.; Simmons, K.B.; Fink, P.J. Recent thymic emigrants and mature naive T cells exhibit differential DNA methylation at key cytokine loci. J. Immunol. 2013, 190, 6180–6186.
- Hosoya, T.; Kuroha, T.; Moriguchi, T.; Cummings, D.; Maillard, I.; Lim, K.C.; Engel, J.D. GATA-3 is required for early T lineage progenitor development. J. Exp. Med. 2009, 206, 2987–3000.
- Wan, Y.Y. GATA3: A master of many trades in immune regulation. Trends Immunol. 2014, 35, 233–242.
- Yagi, R.; Zhu, J.; Paul, W.E. An updated view on transcription factor GATA3-mediated regulation of Th1 and Th2 cell differentiation. Int. Immunol. 2011, 23, 415–420.
- Hu, X.; Schewitz-Bowers, L.P.; Lait, P.J.P.; Copland, D.A.; Stimpson, M.L.; Li, J.J.; Liu, Y.; Dick, A.D.; Lee, R.W.J.; Wei, L. The Bromodomain and Extra-Terminal Protein Inhibitor OTX015 Suppresses T Helper Cell Proliferation and Differentiation. Curr. Mol. Med. 2019, 18, 594–601.
- Zhu, Y.; Zhao, Y.; Zou, L.; Zhang, D.; Aki, D.; Liu, Y.-C. The E3 ligase VHL promotes follicular helper T cell differentiation via glycolytic-epigenetic control. J. Exp. Med. 2019, 216, 1664–1681.
- Yu, B.; Zhang, K.; Milner, J.J.; Toma, C.; Chen, R.; Scott-Browne, J.P.; Pereira, R.M.; Crotty, S.; Chang, J.T.; Pipkin, M.E.; et al. Epigenetic landscapes reveal transcription factors that regulate CD8+ T cell differentiation. Nat. Immunol. 2017, 18, 573–582.
- Hu, B.; Jadhav, R.R.; Gustafson, C.E.; Le Saux, S.; Ye, Z.; Li, X.; Tian, L.; Weyand, C.M.; Goronzy, J.J. Distinct Age-Related Epigenetic Signatures in CD4 and CD8 T Cells. Front. Immunol. 2020, 11, 585168.
- Karantanos, T.; Christofides, A.; Bardhan, K.; Li, L.; Boussiotis, V.A. Corrigendum: Regulation of T Cell Differentiation and Function by EZH2. Front. Immunol. 2016, 7, 346.
- Stairiker, C.J.; Thomas, G.D.; Salek-Ardakani, S. EZH2 as a Regulator of CD8+ T Cell Fate and Function. Front. Immunol. 2020, 11, 593203.
- Grewal, I.S.; Foellmer, H.G.; Grewal, K.D.; Xu, J.; Hardardottir, F.; Baron, J.L.; Janeway, C.A., Jr.; Flavell, R.A. Requirement for CD40 ligand in costimulation induction, T cell activation, and experimental allergic encephalomyelitis. Science 1996, 273, 1864–1867.
- Tsuchiya, Y.; Naito, T.; Tenno, M.; Maruyama, M.; Koseki, H.; Taniuchi, I.; Naoe, Y. ThPOK represses CXXC5, which induces methylation of histone H3 lysine 9 in Cd40lg promoter by association with SUV39H1: Implications in repression of CD40L expression in CD8+ cytotoxic T cells. J. Leukoc. Biol. 2016, 100, 327–338.
- Kallies, A.; Xin, A.; Belz, G.T.; Nutt, S.L. Blimp-1 transcription factor is required for the differentiation of effector CD8+ T cells and memory responses. Immunity 2009, 31, 283–295.
- Li, Y.; Deng, C.; Hu, X.; Patel, B.; Fu, X.; Qiu, Y.; Brand, M.; Zhao, K.; Huang, S. Dynamic interaction between TAL1 oncoprotein and LSD1 regulates TAL1 function in hematopoiesis and leukemogenesis. Oncogene 2012, 31, 5007–5018.
- Hayami, S.; Kelly, J.D.; Cho, H.S.; Yoshimatsu, M.; Unoki, M.; Tsunoda, T.; Field, H.I.; Neal, D.E.; Yamaue, H.; Ponder, B.A.; et al. Overexpression of LSD1 contributes to human carcinogenesis through chromatin regulation in various cancers. Int. J. Cancer 2011, 128, 574–586.
- Fu, X.; Zhang, P.; Yu, B. Advances toward LSD1 inhibitors for cancer therapy. Future Med. Chem. 2017, 9, 1227–1242.
- Muthuswamy, R.; Berk, E.; Junecko, B.F.; Zeh, H.J.; Zureikat, A.H.; Normolle, D.; Luong, T.M.; Reinhart, T.A.; Bartlett, D.L.; Kalinski, P. NF-κB hyperactivation in tumor tissues allows tumor-selective reprogramming of the chemokine microenvironment to enhance the recruitment of cytolytic T effector cells. Cancer Res. 2012, 72, 3735–3743.
- Loo Yau, H.; Bell, E.; Ettayebi, I.; De Almeida, F.C.; Boukhaled, G.M.; Shen, S.Y.; Allard, D.; Morancho, B.; Marhon, S.A.; Ishak, C.A.; et al. DNA hypomethylating agents increase activation and cytolytic activity of CD8+ T cells. Mol. Cell 2021, 81, 1469–1483.e8.
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499.
- Alfei, F.; Kanev, K.; Hofmann, M.; Wu, M.; Ghoneim, H.E.; Roelli, P.; Utzschneider, D.T.; Von Hoesslin, M.; Cullen, J.G.; Fan, Y.; et al. TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature 2019, 571, 265–269.
- Henning, A.N.; Roychoudhuri, R.; Restifo, N.P. Epigenetic control of CD8+ T cell differentiation. Nat. Rev. Immunol. 2018, 18, 340–356.
- Philip, M.; Fairchild, L.; Sun, L.; Horste, E.L.; Camara, S.; Shakiba, M.; Scott, A.C.; Viale, A.; Lauer, P.; Merghoub, T.; et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 2017, 545, 452–456.
- Peirs, S.; Van der Meulen, J.; Van de Walle, I.; Taghon, T.; Speleman, F.; Poppe, B.; Van Vlierberghe, P. Epigenetics in T-cell acute lymphoblastic leukemia. Immunol. Rev. 2015, 263, 50–67.
More