Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Zhixian Chen and Version 2 by Beatrix Zheng.
Iron is essential for cell survival, and iron deficiency is a known risk factor for many reproductive diseases. Paradoxically, such disorders are also more common in cases of iron overload. Here, the reswearchers evaluated the role of ferroptosis in women’s health, particularly focusing on pre-eclampsia (PE). PE is a multisystem disorder and is one of the leading causes of maternal and perinatal morbidity and mortality, especially when the condition is of early onset. Nevertheless, the exact etiological mechanism of PE remains unclear. Interestingly, ferroptosis, as a regulated iron-dependent cell death pathway, involves a lethal accumulation of lipid peroxides and shares some characteristics with PE pathophysiology.
- ferroptosis
- pre-eclampsia
- iron
- lipid peroxidation
- ferroptotic regulators
1. Ferroptosis and Pre-Eclampsia
1.1. The Role of Iron in Pre-Eclampsia (PE) Pathology
During pregnancy, maternal iron requirements will increase by about 30% to support maternal and infant hematopoiesis, and iron loss is reduced due to, for example, cessation of menstruation during pregnancy. Therefore, pregnant women may have a decreased ability for feedback systems to react to ambient iron exposure, resulting in vulnerability to clinical diseases caused by excess iron [1][106]. As mentioned above, the hepcidin-ferroportin cycle is a crucial mechanism for the regulation of iron homeostasis. Hepcidin restricts iron transport through ferroportin and reduces plasma iron concentrations. Therefore, in healthy pregnancies, the expression level of hepcidin is reduced to increase the absorption of iron in the intestinal tract and the mobilization of stored iron in the liver and spleen, so as to meet the body’s increased iron requirements during pregnancy. To summarize, with the progress of pregnancy, the expression of hepcidin decreases, dietary iron absorption increases, and stored iron release increases. Since the concentrations of hepcidin are inversely connected to the transfer of iron across the placenta, iron transfer to the fetus also increases accordingly [2][107]. In addition, maternal levels of ferritin and transferrin-saturation decrease, while TfR levels increase during the second and third trimesters of normal pregnancy [1][106]. However, in PE, levels of free iron, ferritin, and transferrin-saturation increase, while TfR levels decrease [3][4][5][108,109,110]. However, some studies have shown a decrease in the level of hepcidin in PE patients [6][111], while others have found an elevated hepcidin level in early pregnancy or a normal level in late pregnancy [7][112]. It has been reported that an elevated serum hepcidin level in early pregnancy is associated with PE occurrence [7][112]. So far, current studies on hepcidin level changes in PE patients are controversial, and further studies are needed to elucidate the mechanism.
Moreover, iron levels were significantly correlated with disease severity, and serum iron and ferritin levels in EOPE were distinctly higher than those in LOPE and control groups [8][113]. It has also been shown that, in 18% of PE patients, the transferrin saturation levels are high, which is related to iron overload [9][114]. During normal pregnancy, the maternal plasma blood volume increases at the first trimester and can increase by 30–50% at the third trimester [10][115]. This hypervolemia has little effect on pregnancy complications, while increased plasma volume can cause changes in iron concentration and other factors, which have different effects on normal pregnancy and PE pregnancy [11][116]. Therefore, the abundance of iron in trophoblasts can lead to iron overload, resulting in ferroptosis, to which human trophoblast cells are sensitive, leading to macro-blebbing and vesiculation of the plasma membrane, further resulting in placental dysfunction and trophoblast injury [12][117]. Total Hb levels in maternal blood also fluctuate throughout pregnancy, and both low and high levels are linked to adverse pregnancy outcomes. The likelihood of adverse birth outcomes is inversely correlated with maternal hemoglobin concentration or iron status during pregnancy. High Hb levels, however, as well as high iron levels, appear to be linked to less favorable pregnancy outcomes [1][106]. Similarly, in a randomized controlled trial, Ziaei et al. [13][118] found that women who had higher mean hemoglobin concentrations in the third trimester of pregnancy had an increased risk of hypertension (2.7% vs. 0.8%, p < 0.05), as well as an increased risk of giving birth to small-for-gestational-age (SGA) infants (15.7% vs. 10.3%, p = 0.035).
Obesity is a risk factor of PE and has been relevant to increased inflammation due to elevated levels of interleukin-6 (IL-6), which is a vital regulator of hepcidin and thus may be associated with iron dysregulation [14][119]. Women with PE showed a marked inflammatory response and significantly increased levels of IL-6 and tumor necrosis factor-α (TNF-α) [15][120]. Race is another influencing factor of PE. Mutations in the regulatory gene HFE, which regulates iron homeostasis in vivo, are involved in the regulation of hepcidin expression, leading to hemochromatosis or excessive iron storage, with the two most common mutations (C282Y and H63D) occurring in 14% and 29% of the European White populations, respectively [16][121]. In addition, it has been reported that a gene mutation in ferroportin (Q248H) leads to partial resistance to hepcidin-induced differentiation, with a prevalence of up to 13% in Africans [17][122]. To support the normal increase of erythropoiesis in late pregnancy, the first and third trimesters see nearly a doubling of EPO levels [1][106]. However, women with PE had lower levels of EPO at the third trimester compared to normal pregnancies [6][111]. Interestingly, although total serum transferrin levels of PE patients increased during pregnancy compared with non-pregnancy, they were still much lower than normal pregnancy [18][123]. Moreover, iron supplementation during pregnancy in iron-deficient women may inhibit maternal hepcidin production and/or activity [19][124], causing a continuation of dietary iron absorption, an increase in hemoglobin levels, a rise in blood viscosity, and finally a reduction in placental blood flow [13][118]. Furthermore, excessive dietary iron intake may cause elevated circulating levels of postprandial non-transferrin bound iron (NTBI), resulting in oxidative stress, lipid peroxidation, and the DNA damage of placental cells, which affect placental function [20][125]. It has also been found that miR-30b-5p in PE models can reduce the expression of ferroportin 1 by downregulating Cys2/glutamate antiporter and PAX3, leading to a decrease in GSH and an increase in labile Fe2+, therefore promoting the occurrence of ferroptosis in PE patients [21][126]. The expression of miR-210 was also found to be increased in placenta in PE models, resulting in iron accumulation and autophagosome formation in trophoblast cells and hemosiderin deposition in placental stroma trophoblasts [22][127]. Genetic studies have also found that, in PE, the expression levels of genes mainly responsible for iron metabolism (FTH1 and FTL) are downregulated, leading to disruption of iron uptake and intracellular storage, thereby promoting cellular ferroptosis [23][128].
1.2. The Role of Oxidative Stress in PE Pathology
Until 8–10 weeks of gestation, when the embryo is in a hypoxic and hypoglycemia environment, the maternal spiral arteries are fully obstructed by clots of endothelial cells and blood. The spiral arteries are completely canalized by 10–12 weeks of pregnancy, maternal blood rushes into the placental lacunae, and the fetal villi are exposed to glucose, oxygen, and iron for the first time. Inadequate remodeling of the maternal spiral arteries and shallow EVCT endovascular invasion, which are the pathological features of PE, can result from such rapid perfusion, which can also cause a significant amount of oxidative stress and tissue injury. These outcomes include lipid peroxidation of cell membranes and excessive ferroptosis at the maternal–fetal interface, mainly in trophoblast cells [24][28]. Poorly remodeled spiral arteries pose risks of placental underperfusion, high velocity, and turbulent blood flow, resulting in placental ischemia [25][129] and oxidative stress [26][130], which will cause damage to the placental villi and lead to abnormal levels of angiogenic proteins in maternal blood [27][131]. Excessive secretion of anti-angiogenic factors leads to vascular inflammation, endothelial dysfunction, and maternal vascular damage [26][130], which ultimately results in clinical manifestations of hypertension and multiple maternal organ damage. The production of anti-angiogenic factors increased, such as soluble fms-like tyrosine kinase-1 (sFlt-1) [28][132], a protein that binds to the functional receptor of vascular endothelial growth factor (VEGF), and soluble endoglin (sENG) [29][133]. However, the release of angiogenic placental growth factor (PIGF) is inhibited [30][134], resulting in an imbalance among them. Therefore, this two-stage paradigm of early placental dysplasia coupled with severe maternal organ damage and systemic endothelial dysfunction, first proposed in 1993 [31][135], could be a possible model for determining the pathogenesis of PE (Figure 13).

Figure 13. The two-stage model of PE pathogenesis. Image was created with BioRender.com (accessed on 25 June 2022). VEGF: vascular endothelial growth factor; PIGF: placental growth factor; sFlt-1: soluble fms-like tyrosine kinase-1; sENG: soluble endoglin; ROS: reactive oxygen species; IL-6: interleukin-6; TNF-α: tumor necrosis factor-α.
As the most mutagenic product of lipid peroxidation [32][50], as well as an oxidation product that may lead to ferroptosis [33][136], serum malondialdehyde (MDA) levels have been shown to be significantly elevated in PE and eclampsia patients in multiple studies [34][35][36][137,138,139]. In metabolomic analysis of placental mitochondria, PUFA levels and other mitochondrial abnormalities were significantly higher in patients with severe PE than in controls with normal blood pressure [37][140]. Additionally, histological examinations of the spiral artery walls in many PE patients reveal a buildup of “foam cells” (macrophages harboring low-density lipoproteins) that are lipid-filled, similar to the early stages of atherosclerosis. Although such abnormalities have been observed for decades, the mechanisms behind this type of acute atherosclerosis are largely unknown. One of the key factors might be ferroptosis with lipid peroxidation [38][141]. Moreover, Alahari et al. [39][142] proposed that PE is caused by chronic hypoxia and iron homeostasis disorders at the maternal–fetal interface. Gene expression levels of the von Hippel Lindau (VHL) protein, a key executor of the cellular hypoxia response in PE, are regulated by histone demethylase JMJD6 (Jumonji domain containing protein 6). In PE patients, hypoxia and Fe2+ bioavailability leads to decreased JMJD6 demethylase activity, resulting in downregulated VHL expression, accompanied by changes in placental morphology and reduced pup weights. Moreover, multiple experiments have shown that SOD and GSH-Px activities are significantly decreased, catalase activity is increased, and lipid peroxidation and thromboxane (TX) secretion are increased in the placenta of women with PE [40][41][42][143,144,145]. Vaughan et al. [43][146] exposed the ED27 trophoblast cell line to an oxidizing solution that was rich in Fe2+ and linoleic acid, causing the same changes as in the placenta of women with PE, and these changes were prevented by Fe2+ chelation in the oxidizing solution, except for TX, suggesting that oxidative stress combined with elevated maternal circulating lipids and Fe2+ levels may lead to placental oxidative stress, abnormal placental antioxidants, and TX in PE. The expression profile and function of ferroptosis-related genes (FRGs) in PE also showed that the HIF1α and MAPK8 genes were downregulated and that the PLIN2 gene was upregulated [23][128]. HIF1α is the main transcriptional regulator of hypoxia response and regulates cell survival during stress response. It can also reduce fatty acid β-oxidation and promote lipid storage [44][45][147,148]. The expression of hypoxia-induced genes such as erythropoietin, vascular endothelial growth factor (VEGF), and nitric oxide (NO) synthase is regulated by HIF1α and HIF2α, which are by-products of related oxygen sensing pathways. The expression of HIF1α in the human placenta increases in early gestation and decreases at around 9 weeks, when fetal circulation and oxygenation increase [46][149]. In PE, HIF1α and HIF2α are overexpressed in the placenta and cannot be downregulated during oxygenation [47][150]. Thus, HIFlα appears to be the pathogenic mediator of PE. Likewise, as a member of the mitogen-activated protein kinase (MAPK) family, MAPK8 is activated by environmental stressors and participates in the regulation of multiple signaling pathways, which plays a crucial role in many processes of cell functions [48][151]. Adipogenic differentiation-related protein (ADRP), also known as perisidine 2 (PLIN2), can be enclosed in lipid droplets together with phospholipids to take part in neutral lipid storage [49][152]. Furthermore, advanced oxidation protein products (AOPPs), as novel markers of oxidative stress, have been shown to cause trophoblast cell damage and dysfunction and play a role as a new pathogenic agent in PE. Clinical trials also showed significant differences in mean AOPP levels among normotensive pregnant women and pregnant women with severe or mild PE, and plasma AOPP level was positively correlated with 24 h proteinuria excretion and cystatin C [50][153]. It has also been proved that, in trophoblast cells, ROSs were shown to promote miR-335-5p expression in a p53-dependent manner, further reducing the expression of specific protein 1 (Sp1) and thus inhibiting the transition and migration of epithelial cells to mesenchymal cells [51][154]. The discoveries of these new targets have provided new ideas and evidence for the etiology of PE.
1.3. The Role of Other Ferroptosis Regulators in PE Pathology
Boutet et al. [52][155] found that heme oxygenase-1 (HO-1) and HSP-70 mRNA expression in whole blood was dramatically elevated during fetal and maternal circulations. Blood levels of GPX4 mRNA in the PE group were 1.6 times higher than that in the normal pregnancy group, which suggest that PE is correlated with specific antioxidant responses in maternal and fetal circulation, possibly associated with harmful oxidative stress responses observed in the syndrome. Moreover, genotyping studies also showed that there were significant statistical differences in genotype and allelic frequencies of rs713041 in GPX4 between PE patients and the normal pregnancy group, and the C allele had a higher risk for the pathogenesis of PE. At the same time, the rs713041 genotype was also found to be associated with mild, severe, and early-onset PE. Therefore, rs713041 in GPX4 may play a vital role in the pathogenesis of PE [53][33].
The nuclear factor erythroid-2-related factor 2 (Nrf2), a major regulator of antioxidant response, has recently been shown to prevent ferroptosis (Figure 21k) [54][156]. Wang et al. [55][157] found that Fe2+ content was upregulated and the expressions of SLC7A11, GPX4, and FPN1 were downregulated in PE patients. Hypoxia can promote the translocation of Nrf2 to the nucleus, leading to the activation of the Nrf2/HO-1 signaling pathway. Hypoxia-induced Nrf2 overexpression can inhibit the levels of GSH, MDA, ROSs, and Fe2+ and can promote the activation of the Nrf2/HO-1 signaling pathway and the expressions of SLC7A11, GPX4, and FPN1, which indicate that Nrf2 signaling activation could relieve hypoxia and play a protective role in PE. Interestingly, it has been found that DJ-1, as an important sensor of intracellular redox state, its mRNA expression levels were significantly increased in PE patients [56][158]. Its expression level is inversely connected to MDA concentration and favorably correlated with the Nrf2/GPX4 signaling pathway expression levels. In order to protect the body from toxins, DJ-1 can dissociate Nrf2 and Keap-1, translocating Nrf2 into the nucleus where it binds with antioxidant response element (ARE) [57][159]. This increases the expression of a series of downstream antioxidant enzymes, such as GPX4 and SOD. In other words, DJ-1 can control the Nrf2/GPX4 signaling pathway, causing ferroptosis in trophoblast cells and acting as a protective factor in the pathogenesis of PE [58][160].
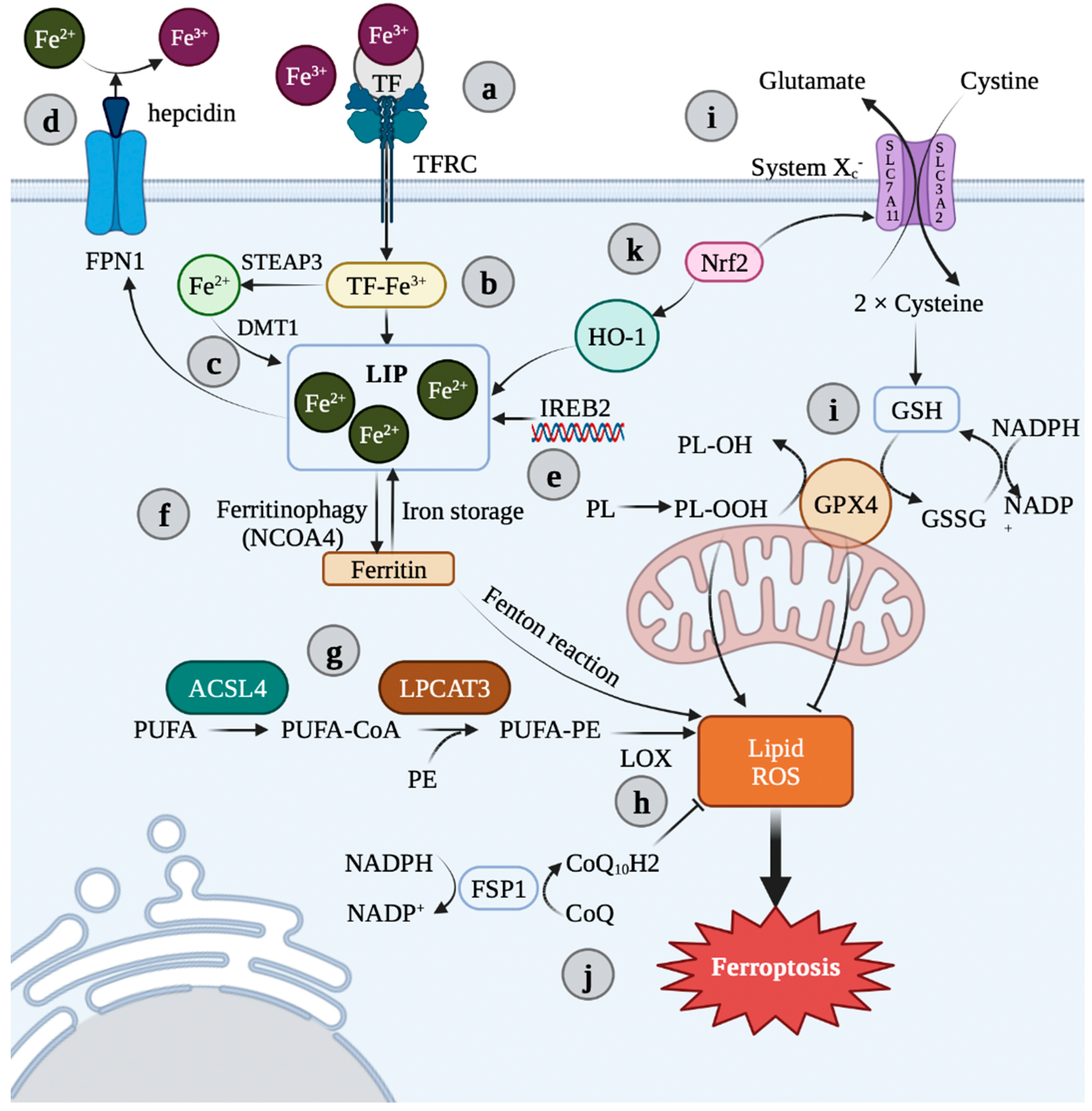
Figure 2. Overview of ferroptosis pathways. (a) TFRC is bound to TF and two Fe3+, which then enter the cell via endocytosis. (b) In endosomes, Fe3+ is reduced to Fe2+ via STEAP3. (c) Fe2+ can then be stored as ferritin or act in an active loose state known as a “liable iron pool” (LIP) by DMT1. (d) Fe2+ can be released into the plasma via ferroportin (FPN), while hepcidin can bind to FPN to inhibit Fe2+ release and regulate iron homeostasis. (e) IREB2 is also a key regulator of iron content. (f) Ferritinophagy and LIP contribute to iron load, and excess iron is the cofactor of LOX. (g) ACSL4 and LPCAT3 are involved in the manufacture and modification of PUFA-PE containing polyunsaturated fatty acid in the cell membrane. (h) LOX (mainly LOX-15) mediates the peroxidation of PUFA-PE to conduct the ferroptosis axis. (i) Ferroptosis is controlled by two major regulatory systems, namely, the transporter system Xc−, which is composed of SLC3A2 and SLC7A11, and the GSH/GPX4. (j) In addition, there are GSH-independent antioxidant pathways, such as the CoQ10 axis. The FSP1 in the plasma membrane showed oxidoreductase activity. It reduces the coenzyme Q and decreases the accumulation of L-OOH. (k) The Nrf2/HO-1 axis can promote the increase of Fe2+ by catabolizing heme. Image was created with BioRender.com (accessed on 25 June 2022). FPN 1: ferroportin 1; TF: transferrin; TFRC: transferrin receptor; LIP: liable iron pool; Nrf2: nuclear erythroid 2-related factor 2; HO-1: heme oxygenase-1; SLC7A11: solute carrier gamily 7 member 11; SLC3A2: solute carrier gamily 3 member 2; IREB2: iron responsive element binding protein 2; GSH: glutathione; GSSG: oxidized glutathione; GPX4: glutathione peroxidase 4; NCOA4: nuclear receptor coactivator 4; ACSL4: Acyl-CoA synthetase long-chain family member 4; LPCAT3: lysophosphatidylcholineacyl transferase 3; PUFA-PE: phosphatidylethanolamine; LOX: lipoxygenase; ROS: reactive oxygen species; FSP1: ferroptosis suppressor protein 1; Fe3+: ferric iron; Fe2+: ferrous iron; STEAP3: six-transmembrane epithelial antigen of prostate 3; DMT1: divalent metal transporter 1.
CoQ10 is essential for energy production and the development of ROSs since it is the only non-polar electron transporter in the mitochondrial respiratory chain [59][161]. Circulating CoQ10, either directly or through the production of vitamin E, is considered as a potential antioxidant [60][162]. Therefore, it makes sense that CoQ10 might contribute to the onset of PE. Initial studies have shown that CoQ10 levels in PE patients rise gradually from the first trimester and continue to rise until delivery [61][163]. However, subsequent studies have shown that CoQ10 levels are significantly downregulated in women with PE [62][164]. A hypothesis was proposed suggesting that there might be a “mechanism” for CoQ10 consumption during PE (independent of diet), possibly due to an increased production of ROSs [63][165]. Intriguingly, the researchers discovered significantly higher amounts of CoQ10 in the placenta and the umbilical cord of women with PE, suggesting compensatory accumulation [64][166]. To control for altitude, several subsequent studies at sea level showed that CoQ10 levels were significantly reduced in normal pregnant women, but the difference was less pronounced in women with PE than in those living at higher altitudes. However, at sea level, CoQ10 levels were also significantly higher in the placentas of women with PE [65][167]. Subsequent clinical trials showed that CoQ10 supplementation was an effective intervention to reduce PE risk [66][168]. In patients with PE, although CoQ10 reduced the incidence of PE, and the levels of CoQ10 in placental tissues were high, the mitochondrial levels of CoQ10 did not change significantly [67][169]. In other words, CoQ10 levels may have varied between healthy pregnant women and PE patients. The plasma and placenta both undergo these modifications, but they are more likely to be found in mitochondria. It is unknown whether the change in the level of CoQ10 in PE patients involves the participation of ferroptosis mechanism, and more research are needed.
2. Ferroptosis and PE Therapy
Because placental hypoxia is the core of PE pathogenesis, it has been a main focus for developing novel treatments for PE. Ouabain, a digoxin-like molecule that can inhibit HIF1 and HIF2, has been shown to block sFlt-1 (a soluble splice variant of the membrane-bound receptor VEGFR1) production and reduce hypertension in placental ischemia rats [68][170]. In addition, antioxidants have been studied for their potential beneficial effects on PE. For example, in a clinical trial of aspirin, treatment started at ≤16 weeks of gestation at a daily dose of ≥100 mg significantly reduced the risk of preterm PE compared with patients who were not treated with aspirin (relative risk: 0.33; 95% confidence interval: 0.19–0.57) [69][171]. Additionally, studies have showed that aspirin treatment at 150 mg per day reduced preterm PE by 62% when compared to the placebo [70][172]. Low-dose aspirin is now recommended for PE prevention in high-risk women, although there is currently a dearth of conclusive evidence proving how aspirin treatment reduces prenatal morbidity and death [71][173]. Studies have also shown that, in high-risk women, the initiation of treatment with low-molecular-weight heparin before 16 weeks of gestation can significantly reduce the risk of PE and other placenta-mediated complications. Combined treatment with low-dose aspirin significantly reduced the risk of PE compared with low-dose aspirin alone [72][73][174,175]. However, non-specific antioxidants, such as the use of vitamin C and vitamin E, have not been shown to be effective in preventing PE in clinical trials [74][75][176,177]. As a result, the causes of oxidative stress in PE patients are receiving increasing amounts of attention in order to further identify therapeutic targets. Mitochondrial oxidative stress has become an attractive target, and the use of mitochondrial-targeted antioxidants as a therapeutic strategy to reverse oxidative stress in PE has become an emerging research topic [76][178]. For example, AP39, a novel mitochondrial-targeted hydrogen sulfide donor, has been reported to block ROS production, reduce HIF-1α protein levels, and reduce sFLT1 production. At the same time, as a mitochondrial bioregulatory factor, it can enhance the activity of cytochrome c oxidase and reverse the oxidative stress and anti-angiogenesis response of hypoxic trophoblast cells [77][179].
In addition, statins are effective in the long-term prevention of cardiovascular diseases and mortality, not only through their lipid-lowering mechanisms but also through their regulation of inflammation, antioxidants, and endothelial homeostasis, among other pleiotropic effects [78][180]. The inhibition of HMG-CoA reductase by statins disrupts membrane synthesis [79][181]. They may also interfere with cell proliferation, growth, and metabolism and protein glycosylation, which all play important roles in normal placental development. Statins may also disrupt the placenta by expressing peroxisome proliferator-activated receptor γ and inhibit trophoblast invasion [80][182]. In a patient with poor obstetric outcomes, Otten et al. [81][183] administered 10 mg of pravastatin daily beginning in the second trimester of pregnancy, achieving a normal pregnancy and full-term delivery of a healthy neonate suitable for gestational age in a patient with a history of severe, early-onset, recurrent HELLP syndrome. There are various findings supporting the use of pravastatin as the statin of choice for the treatment of early and severe PE. Pravastatin, as the most hydrophilic statin, can restrict placental transfer and be metabolized and cleared through liver and kidney pathways. In addition, pravastatin has shown multifactorial activities in sFlt-1-induced mouse PE, e.g., promoting the expression level of VEGF, fueling endothelial function, upregulating vascular eNOS, and stimulating vascular reactivity. The use of statins to treat PE has become dominant in the past few years [80][182].
However, although these agents have been shown to be associated with oxidative stress in the placenta, their specific roles in the regulation of ferroptosis remain unclear. To the best of the ouresearchers' knowledge, currently there is no report on specific treatment of the ferroptosis regulation pathway in PE, which warrants further investigation.
At the same time, excluding oxidative stress, iron overload is another landmark feature of ferroptosis, and its treatment scheme as a target is also a hotspot in ferroptosis research. Areas of interest include the use of iron chelating agents such as desferrioxamine and deferiprone. However, maternal desferrioxamine therapy is mostly used in pregnant women with beta-thalassemia [81][183], and its application in PE pregnant women has not been effectively studied. Therefore, in order to find more effective treatment options for PE patients based on ferroptosis, iron chelating agents require further study.