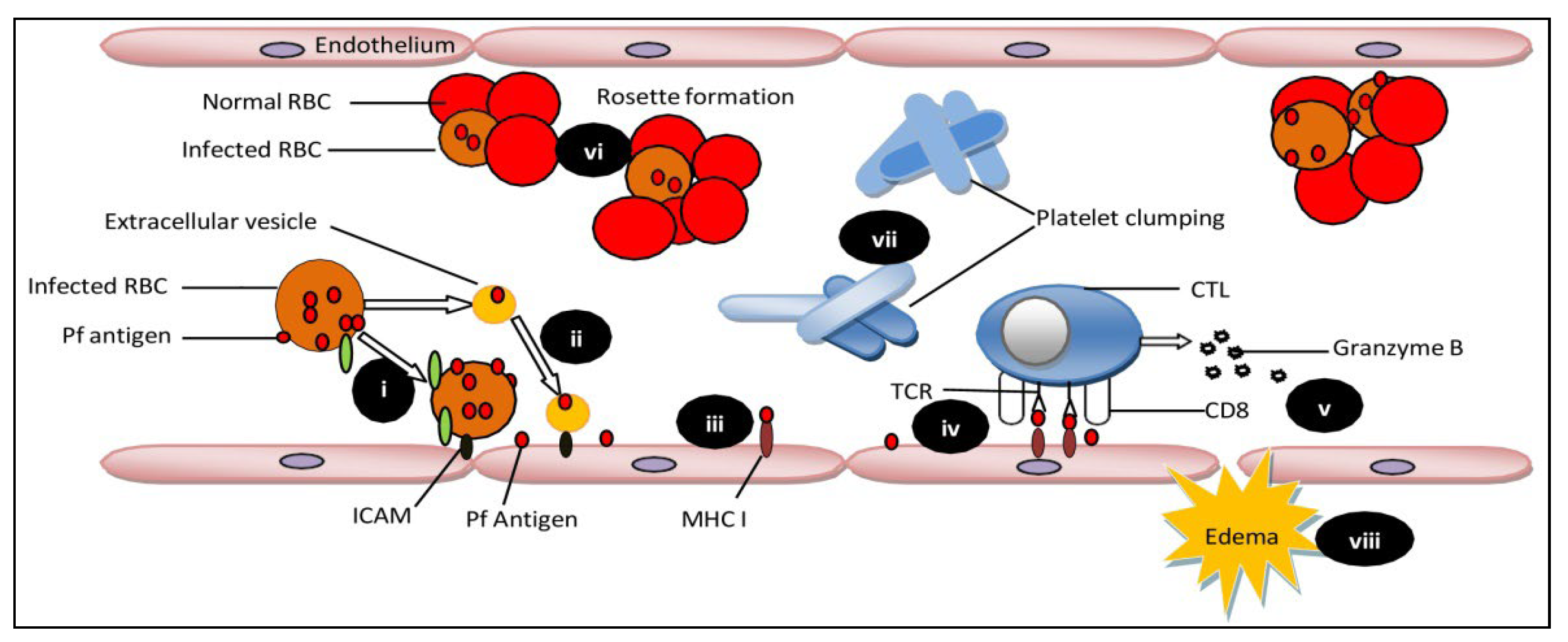
Plasmodium falciparum causes over 90 percent of all malaria infections. Children under the age of 5 years and pregnant women were the most susceptible groups affected by malaria. The World Health Organization (WHO) has characterized malaria as severe and uncomplicated. Delays in the detection and treatment of an uncomplicated infection of P. falciparum malaria lead to complications of severe cerebral malaria (CM). CM is usually caused by P. falciparum, but Plasmodium vivax is rarely responsible for CM complications. CM is a severe neurological complication caused by Plasmodium falciparum infection, resulting in high mortality rates. CM is characterized by brain tissue hemorrhage, the accumulation of infected red blood cells and mononuclear cells in brain microvessels, and blood-brain barrier (BBB) disruption.
- cerebral malaria
- pathophysiology
- cytoadherence
1. Pathophysiology during Cerebral Malaria Progression
2. Therapeutic Approaches
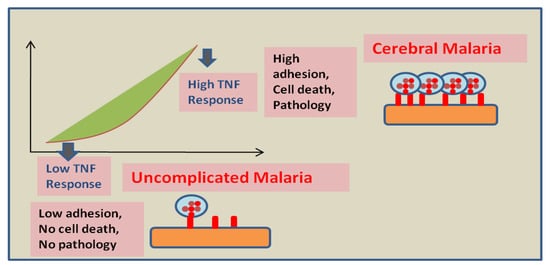
The sequestration of infected RBCs can also be prevented by using drugs targeting cytoadherence on the endothelium. In experimental cerebral malaria reports, the use of rapamycin restricts the cytoadherence of infected RBCs on endothelial cells by VCAM-1 and ICAM-1 reduction [23]. Moreover, the fungal ethanolic extract of Trichoderma stromaticum helps in reducing inflammation in experimental cerebral malaria by reducing the expression of VCAM-1 and ICAM-1, thus protecting the blood-brain barrier’s integrity [24]. The activation of endothelial cells by cytoadherence of infected RBCs and the pro-inflammatory effects of released cytokines play important roles in cerebral malaria pathophysiology [25]. Anti-TNF treatments can be helpful in managing the severity of disease and mortality rate. Endothelium activation triggers a TNF dependent pro-apoptotic pathway [26]. The difference in response of endothelial cells to TNF can affect the severity of the disease [27]. Anti-TNF therapy showed a positive result in vitro. However, it showed no reduction in mortality in the patients. Moreover, the use of antibodies against endothelial protein C receptor inhibited PfEMP1′s ability to bind to human endothelial cells [28]. Further studies on endothelial cells confirm the activation of Rac1 signaling via cross binding of VCAM-1 that results in Rho-dependent inductions of stress fibers, leading to the weakening of tight junctions [29]. Moreover, the interaction of EPCR with activated protein C is prevented by binding PfEMP1 to EPCR. This binding shows two impacts: (i) promoting the activation of tissue factors Va and VIIIa, which results in the disablement of the aPC-mediated anti-coagulative pathway. These factors are responsible for thrombin generation on activation, which eventually results in fibrin deposition. (ii) The initiation of NF-kB and Rho A pathways through thrombin-mediated scissions of PAR1 is also observed. The activation of these pathways generates a pro-inflammatory response, which leads to impairment and breakage of the blood-brain barrier [11][30][31]. The administration of aPC may prevent blood-brain barrier dysfunction by inhibiting thrombin activity [32]. Other approaches embrace the utilization of endothelial cells isolated from patients suffering with cerebral malaria. Moreover, measuring the consequences of Ang-1 on endothelial cells pre-treated with TNF will be crucial for the development of new additional therapies and improving disease outcomes in CM. Since Ang-1 acts as a serenity agent for endothelial cells in the exquisite model of sepsis [33], maintaining a higher Ang-1concetration in Ang-2/Ang-1 ratio during infection can potentially block and reverse ongoing inflammatory processes in CM patients.3. Mesenchymal SCtem Cells as A Regenerative Therapy
3.1. MSCs Mediated Cellular Mechanism of Protection
3.1. Mesenchymal Stem Cells Mediated Cellular Mechanism of Protection
Mesenchymal stem cells (MSCs) first observed in the bone marrow are multipotent cells [34], which can differentiate into various cell lines lineages such as adipocytes, chondrocytes, and osteoblasts and display strong tissue protective and restorative properties. During malaria infection, MSCs become accumulated in primary and secondary lymphoid organs. Different surface antigens are expressed by MSCs in different tissues, which facilitate tissue repair and regeneration through the secretion of various soluble factors. Souza MC et al. demonstrated that, in P. berghei-infected mice, neurons were damaged, with an increased number of astrocytes and oligodendrocytes. However, in P. berghei-infected mice treated with BM-MSCs, brain damage was repaired, which leads to a reduction in parasitemia and mortality [35]. There was a substantial increase in phagocytic neutrophils in the brain [36]; hepatocytes and Kupffer cell regeneration in MSC-infused mice indicate regenerative ability of MSCs. MSCs promote hematopoiesis by releasing different critical molecules involved in the self-renewal, proliferation, and differentiation of hematopoietic stem and progenitor cells (HSPCs). Other than that, MSCs encourage the formation of colony-forming units-erythroid (CFU-E) cells in the bone marrow [37], which ultimately helps in preventing malarial anemia. These observations suggest that cell-based therapeutics for intervention in malaria may be useful in achieving sterile clearance and preventing disease reactivation.3.2. MSCs Mediated Molecular Aspects of Protection
3.2. Mesenchymal Stem Cells Mediated Molecular Aspects of Protection
Studies on anti -CD3−, -CD19−, -Sca-1+, and -CD34+ antibodies revealed a significant elevation in Sca- 1+ and CD34+ cells in the lymph node and spleen of MSC-treated mice as compared to untreated mice [36]. A drop in GATA-1 and GATA-2 expression was observed in plasmodium-infected mice. Since GATA-1 is a key erythroid cell differentiation factor, the low level of GATA-1 expression may be responsible for reduced RBC formation in malaria-infected animals. However, after MSCs treatment, an increase in the expression of GATA-1 and GATA-2 has been reported in animals infused with MSCs. Increased CFU-E formation and reduced hemozoin content in MSC-infused mice support the conclusion that MSCs elicits signals to support erythropoiesis [38].3.3. Application of MSCs in Animal Model of Cerebral Malaria
3.3. Application of Mesenchymal Stem Cells in Animal Model of Cerebral Malaria
Malaria infection causes a reduction in CD4+ T cells in mice models of cerebral malaria, which are crucial for immunity development against malaria, leading to impaired T cell-mediated immunity [39]. However, an increase in the number of CD4+ and CD8+ T cells was reported in MSC-infused mice, and MSCs are able to rescue the proliferation of CD4+ T cells [37]. Ongoing research studies are focused on signaling molecules engaged in restoring immune responses by MSCs. MSCs also inhibited the induction of the negative co-stimulatory receptor programmed death-1 by T cells in recipient animals. Taken together, MSCs help in the protection against malaria infection by reprogramming hematopoiesis, by enhancing the differentiation of CD34+ cells, restoring CD4+ and CD8+ T cell proliferation, and by suppressing the expression of negative co-stimulators on T cells. The increased production of interleukin IL-12, which is crucial for self-renewal and differentiation of multipotent progenitor cells and suppressed production of IL-10, was reported in MSC infused animals. The transfer of MSCs isolated from secondary lymphoid organs of P. berghei-infected mice conferred host resistance against malaria through the enhanced production of pro-inflammatory cytokines IL-6, IL-12, and TNF-α, and the suppression of IL-10 [38][40]. Preclinical studies on several metabolic disorders, tissue regeneration, cancer, heart disorders, and other disorders suggest the robust use of MSC-based therapy [41]. Despite multiple preclinical studies on MSC-based therapy, no promising clinical studies are available for cerebral malaria. The multipotent nature and the potential of MSCs of modifying the tissue microenvironment makes them appropriate candidates for the development of stem cell-based regenerative medicinal therapy.References
- Wassmer, S.C.; Grau, G.E.R. Severe malaria: What’s new on the pathogenesis front? Int. J. Parasitol. 2017, 47, 145–152.
- Kaul, D.; Roth, E.J.; Nagel, R.; Howard, R.; Handunnetti, S. Rosetting of Plasmodium falciparum-infected red blood cells with uninfected red blood cells enhances microvascular obstruction under flow conditions. Blood 1991, 78, 812–819.
- Rowe, J.A.; Claessens, A.; Corrigan, R.A.; Arman, M. Adhesion of Plasmodium falciparum-infected erythrocytes to human cells: Molecular mechanisms and therapeutic implications. Expert Rev. Mol. Med. 2009, 11, e16.
- Wassmer, S.C.; de Souza, J.B.; Frère, C.; Candal, F.J.; Juhan-Vague, I.; Grau, G.E. TGF-β1 released from activated platelets can induce TNF-stimulated human brain endothelium apoptosis: A new mechanism for microvascular lesion during cerebral malaria. J. Immunol. 2006, 176, 1180–1184.
- Prapansilp, P.; Medana, I.; Mai, N.T.H.; Day, N.P.; Phu, N.H.; Yeo, T.W.; Hien, T.T.; White, N.J.; Anstey, N.M.; Turner, G.D. A clinicopathological correlation of the expression of the angiopoietin-Tie-2 receptor pathway in the brain of adults with Plasmodium falciparum malaria. Malar. J. 2013, 12, 1–15.
- Adams, Y.; Olsen, R.W.; Bengtsson, A.; Dalgaard, N.; Zdioruk, M.; Satpathi, S.; Behera, P.K.; Sahu, P.K.; Lawler, S.E.; Qvortrup, K. Plasmodium falciparum erythrocyte membrane protein 1 variants induce cell swelling and disrupt the blood–brain barrier in cerebral malaria. J. Exp. Med. 2021, 218, e20201266.
- Gallego-Delgado, J.; Rodriguez, A. Rupture and Release: A Role for Soluble Erythrocyte Content in the Pathology of Cerebral Malaria. Trends Parasitol. 2017, 33, 832–835.
- Griffith, J.W.; Sun, T.; McIntosh, M.T.; Bucala, R. Pure Hemozoin is inflammatory in vivo and activates the NALP3 inflammasome via release of uric acid. J. Immunol. 2009, 183, 5208–5220.
- Rénia, L.; Grau, G.E.; Wassmer, S.C. CD8+ T cells and human cerebral malaria: A shifting episteme. J. Clin. Investig. 2020, 130, 1109–1111.
- Carman, C.V.; Jun, C.-D.; Salas, A.; Springer, T.A. Endothelial cells proactively form microvilli-like membrane projections upon intercellular adhesion molecule 1 engagement of leukocyte LFA-1. J. Immunol. 2003, 171, 6135–6144.
- Mosnier, L.O.; Zlokovic, B.V.; Griffin, J.H. The cytoprotective protein C pathway. Blood 2007, 109, 3161–3172.
- Bernabeu, M.; Smith, J.D. EPCR and malaria severity: The center of a perfect storm. Trends Parasitol. 2017, 33, 295–308.
- Moxon, C.A.; Wassmer, S.C.; Milner Jr, D.A.; Chisala, N.V.; Taylor, T.E.; Seydel, K.B.; Molyneux, M.E.; Faragher, B.; Esmon, C.T.; Downey, C. Loss of endothelial protein C receptors links coagulation and inflammation to parasite sequestration in cerebral malaria in African children. Blood J. Am. Soc. Hematol. 2013, 122, 842–851.
- Hunt, N.H.; Grau, G.E. Cytokines: Accelerators and brakes in the pathogenesis of cerebral malaria. Trends Immunol. 2003, 24, 491–499.
- Grau, G.E.; Fajardo, L.F.; Piguet, P.-F.; Allet, B.; Lambert, P.-H.; Vassalli, P. Tumor necrosis factor (cachectin) as an essential mediator in murine cerebral malaria. Science 1987, 237, 1210–1212.
- Jambou, R.; Combes, V.; Jambou, M.-J.; Weksler, B.B.; Couraud, P.-O.; Grau, G.E. Plasmodium falciparum adhesion on human brain microvascular endothelial cells involves transmigration-like cup formation and induces opening of intercellular junctions. PLoS Pathog. 2010, 6, e1001021.
- Wassmer, S.C.; Taylor, T.E.; Rathod, P.K.; Mishra, S.K.; Mohanty, S.; Arevalo-Herrera, M.; Duraisingh, M.T.; Smith, J.D. Investigating the Pathogenesis of Severe Malaria: A Multidisciplinary and Cross-Geographical Approach. Am. J. Trop. Med. Hyg. 2015, 93 (Suppl. S3), 42–56.
- El-Assaad, F.; Wheway, J.; Mitchell, A.J.; Lou, J.; Hunt, N.H.; Combes, V.; Grau, G.E.R. Cytoadherence of Plasmodium berghei-infected red blood cells to murine brain and lung microvascular endothelial cells in vitro. Infect. Immun. 2013, 81, 3984–3991.
- Chang, K.-H.; Stevenson, M.M. Malarial anaemia: Mechanisms and implications of insufficient erythropoiesis during blood-stage malaria. Int. J. Parasitol. 2004, 34, 1501–1516.
- Chasis, J.A.; Mohandas, N. Erythroblastic islands: Niches for erythropoiesis. Blood J. Am. Soc. Hematol. 2008, 112, 470–478.
- Borggrefe, T.; Waskow, C.; Roeder, R.G.; Stumpf, M. Severely Impaired Erythropoiesis in Mice Lacking Mediator Subunit Med1/TRAP220. Am. Soc.Hematol. 2004, 104, 1611.
- Tang, Y.; Joyner, C.J.; Cabrera-Mora, M.; Saney, C.L.; Lapp, S.A.; Nural, M.V.; Pakala, S.B.; DeBarry, J.D.; Soderberg, S.; Kissinger, J.C. Integrative analysis associates monocytes with insufficient erythropoiesis during acute Plasmodium cynomolgi malaria in rhesus macaques. Malar. J. 2017, 16, 384.
- Mejia, P.; Treviño-Villarreal, J.H.; Reynolds, J.S.; De Niz, M.; Thompson, A.; Marti, M.; Mitchell, J.R. A single rapamycin dose protects against late-stage experimental cerebral malaria via modulation of host immunity, endothelial activation and parasite sequestration. Malar. J. 2017, 16, 455.
- Cariaco, Y.; Lima, W.R.; Sousa, R.; Nascimento, L.A.C.; Briceño, M.P.; Fotoran, W.L.; Wunderlich, G.; Dos Santos, J.L.; Silva, N.M. Ethanolic extract of the fungus Trichoderma stromaticum decreases inflammation and ameliorates experimental cerebral malaria in C57BL/6 mice. Sci Rep. 2018, 8, 1547.
- Moxon, C.A.; Heyderman, R.S.; Wassmer, S.C. Dysregulation of coagulation in cerebral malaria. Mol. Biochem. Parasitol. 2009, 166, 99–108.
- Wassmer, S.C.; Combes, V.; Grau, G.E. Pathophysiology of Cerebral Malaria. Ann. N. Y. Acad. Sci. 2003, 992, 30–38.
- Combes, V.; Coltel, N.; Faille, D.; Wassmer, S.C.; Grau, G.E. Cerebral malaria: Role of microparticles and platelets in alterations of the blood–brain barrier. Int. J. Parasitol. 2006, 36, 541–546.
- Avril, M.; Bernabeu, M.; Benjamin, M.; Brazier, A.J.; Smith, J.D. Interaction between Endothelial Protein C Receptor and Intercellular Adhesion Molecule 1 to Mediate Binding of Plasmodium falciparum-Infected Erythrocytes to Endothelial Cells. mBio 2016, 7, e00615-16.
- Wittchen, E.S. Endothelial signaling in paracellular and transcellular leukocyte transmigration. Front. Biosci. 2009, 14, 2522.
- Kessler, A.; Dankwa, S.; Bernabeu, M.; Harawa, V.; Danziger, S.A.; Duffy, F.; Kampondeni, S.D.; Potchen, M.J.; Dambrauskas, N.; Vigdorovich, V.; et al. Linking EPCR-Binding PfEMP1 to Brain Swelling in Pediatric Cerebral Malaria. Cell Host Microbe 2017, 22, 601–614.e605.
- Roy, R.V.; Ardeshirylajimi, A.; Dinarvand, P.; Yang, L.; Rezaie, A.R. Occupancy of human EPCR by protein C induces β-arrestin-2 biased PAR1 signaling by both APC and thrombin. Blood 2016, 128, 1884–1893.
- Glennon, E.K.K.; Dankwa, S.; Smith, J.D.; Kaushansky, A. Opportunities for Host-targeted Therapies for Malaria. Trends Parasitol. 2018, 34, 843–860.
- Mei, S.H.J.; McCarter, S.D.; Deng, Y.; Parker, C.H.; Liles, W.C.; Stewart, D.J. Prevention of LPS-induced acute lung injury in mice by mesenchymal stem cells overexpressing angiopoietin 1. PLoS Med. 2007, 4, e269.
- Kalkal, M.; Tiwari, M.; Thakur, R.S.; Awasthi, V.; Pande, V.; Chattopadhyay, D.; Das, J. Mesenchymal Stem Cells: A Novel Therapeutic Approach to Enhance Protective Immunomodulation and Erythropoietic Recovery in Malaria. Stem Cell Rev. Rep. 2021, 17, 1993–2002.
- Souza, M.C.; Silva, J.D.; Pádua, T.A.; Torres, N.D.; Antunes, M.A.; Xisto, D.G.; Abreu, T.P.; Capelozzi, V.L.; Morales, M.M.; Sa Pinheiro, A.A. Mesenchymal stromal cell therapy attenuated lung and kidney injury but not brain damage in experimental cerebral malaria. Stem Cell Res. Ther. 2015, 6, 102.
- Chen, L.; Zhang, Z.; Sendo, F. Neutrophils play a critical role in the pathogenesis of experimental cerebral malaria. Clin. Exp. Immunol. 2000, 120, 125–133.
- Thakur, R.S.; Awasthi, V.; Sanyal, A.; Chatterjee, S.; Rani, S.; Chauhan, R.; Kalkal, M.; Tiwari, M.; Pande, V.; Das, J. Mesenchymal stem cells protect against malaria pathogenesis by reprogramming erythropoiesis in the bone marrow. Cell Death Discov. 2020, 6, 125.
- Thakur, R.S.; Tousif, S.; Awasthi, V.; Sanyal, A.; Atul, P.; Punia, P.; Das, J. Mesenchymal stem cells play an important role in host protective immune responses against malaria by modulating regulatory T cells. Eur. J. Immunol. 2013, 43, 2070–2077.
- Xu, H.; Wipasa, J.; Yan, H.; Zeng, M.; Makobongo, M.O.; Finkelman, F.D.; Kelso, A.; Good, M.F. The mechanism and significance of deletion of parasite-specific CD4+ T cells in malaria infection. J. Exp. Med. 2002, 195, 881–892.
- Hansen, D.S.; Schofield, L. Natural regulatory T cells in malaria: Host or parasite allies? PLoS Pathog. 2010, 6, e1000771.
- Schäfer, R.; Spohn, G.; Baer, P.C. Mesenchymal stem/stromal cells in regenerative medicine: Can preconditioning strategies improve therapeutic efficacy. Transfus. Med. Hemotherapy 2016, 43, 256–267.