Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 3 by Catherine Yang and Version 2 by Catherine Yang.
Cardiotoxicity has emerged as a major side effect of doxorubicin (DOX) treatment, affecting nearly 30% of patients within 5 years after chemotherapy. Heart failure is the first non-cancer cause of death in DOX-treated patients. Although many different molecular mechanisms explaining the cardiac derangements induced by DOX were identified in past decades, the translation to clinical practice has remained elusive to date.
- DOX
- anthracycline
- cardiomyopathy
- doxorubicin
- mitochondrial dysfunction
1. Cell Death
Mitochondrial dysfunction was the first observed mechanism associated with cardiomyocyte toxicity induced by DOX. In fact, Doroshow and Davies reported in the ’80s that DOX generates superoxide anion and ROS in bovine submitochondrial heart preparations, which in turn contribute to the derangement of the mitochondrial electron transport chain (ETC), particularly complex I [1][2]. The specificity of DOX for the heart was explained by its high affinity for cardiolipin, a phospholipid particularly abundant in the inner mitochondrial membrane (IMM), and by the elevated mitochondrial mass that characterizes the heart to meet its energy demand [3]. DOX irreversibly binds cardiolipin, impairing mitochondrial membrane integrity and subtracting a crucial anchor point for cytochrome C [4]. The latter triggers a harmful radical chain reaction that culminates in apoptosis [5]. The importance of this interaction was also confirmed by treating with DOX cardiolipin-deficient human lymphocytes harvested from patients suffering from Barth’s syndrome, a multiorgan genetic disease originating from the mutation of Tafazzin (TAZ), an essential protein for cardiolipin biosynthesis. These cells display resistance to DOX-induced damage compared to those derived from healthy patients [6]. In addition, H9C2 cardiac cells silenced for TAZ show reduced DOX-induced oxidative damage [6]. These results suggest that cardiolipin represents an important target in DOX-induced cardiotoxicity.
Despite being generally associated with a protective cardiovascular effect, endothelial nitric oxide synthase (eNOS) also appears to play an essential role in mediating the acute cardiotoxic effects of DOX. eNOS-KO mice treated with a single dose of DOX (20 mg/kg) show preserved cardiac function and reduced cell death compared to wild-type and transgenic eNOS overexpressing animals [7]. However, mice receiving fenofibrate display preserved cardiac function and structure through the activation of the Akt/eNOS pathway in response to chronic administration of the same cumulative dose of DOX (20 mg/kg), whereas pharmacological eNOS inhibition through L-NAME abolishes the protective effects of fenofibrate [8]. These studies prove that acute and chronic DCM may be characterized by differential molecular signatures underlying the development of cardiac dysfunction.
Besides cardiolipin, increased cardiomyocyte ROS production was also associated with mitochondrial iron overload in human heart specimens from patients suffering from DCM but not from other cardiomyopathies [9]. Of note, cardiac-specific overexpression of ABCB8, a mitochondrial exporter of iron, markedly reduces ROS production and myocardial dysfunction in a murine model of DCM [9]. Marked iron accumulation eventually leads to cardiomyocyte ferroptosis, a peculiar form of non-apoptotic programmed cell death involving lipid peroxidation [10]. In several independent studies, ferroptotic cell death was countered by administering dexrazoxane, which improves redox status and preserves myocardial function in murine preclinical models of DOX treatment using mice and rats [9][11][12]. Dexrazoxane is FDA-approved for the cardioprotection of oncological patients and was successfully used in pediatric patients with acute lymphoblastic leukemia [13] and, to date, is being tested in breast cancer patients in the PHOENIX1 phase I clinical trial (NCT03930680).
Together with ferroptosis, DOX accumulation may lead to a high-conductance mitochondrial permeability transition pore (mPTP) opening [14], a phenomenon that releases mitochondrial Ca2+ and Cytochrome C contributing to apoptotic or necrotic cell death, depending on the nature of the upstream stress [15][16]. Cyclosporin-A (CyA), an mPTP opening inhibitor, was administered to mice chronically receiving DOX and was proven to protect myocardial function and reduce cardiomyocyte mortality after 16 weeks [17]. Mitochondrial-dependent necrotic cell death was found to be linked to DOX-induced repression of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), which leads to increased levels of the mitochondrial B-cell lymphoma 2 (BCL-2)-Interacting Protein 3 (BNIP-3) [18][19]. Systemic BNIP-3−/− mice treated with DOX show normal heart function, absence of myocardial remodeling, and preserved mitochondrial respiration, confirming a prominent role for this protein in DCM [20]. The cardiotoxic induction of BNIP-3 in response to DOX was also reduced in mice overexpressing translationally controlled tumor protein (TCTP), which is downregulated in response to DOX, thereby triggering cardiomyocyte death and heart dysfunction [21]. In addition, chronic DOX-induced contractile dysfunction and cardiomyocyte apoptosis are reduced in p53+/− and in mice with cardiac overexpression of BCL-2 by impairing the oxidative DNA damage/Ataxia telangiectasia mutated (ATM)/p53 pro-apoptotic pathway [22].
Together, these studies indicate that inhibiting pro-apoptotic pathways protects the heart from developing DCM in animal models.
2. Metabolic Derangements
Besides cell death mechanisms, also mitochondrial metabolism is impaired in DOX-induced cardiotoxicity. In metabolomic studies conducted on rats, disorders with energy metabolism, fatty acids oxidation, amino acids, purine and choline metabolism, and gut microbiota-related metabolism were all associated with DOX administration [23]. Treated rats display a reduction in crucial mitochondrial elements, including superoxide dismutase (SOD) and nuclear respiratory factor 2 (NRF-2), which mediate stress response, and protein tyrosine phosphatase 1B (PTP1B), whose activation impairs insulin receptor substrate 1 (IRS-1), leading to disorders of fatty acid metabolism and glycolysis [24]. Aldose reductase promotes high glucose-induced superoxide overproduction insult. Its inhibition with fidarestat preserved heart function, reduced circulating troponin I, and increased mitochondrial biogenesis in nude mice receiving colorectal cancer xenografts and treated with DOX [25][26]. This effect might be linked to the activation of the Sirtuin 1—Peroxisome proliferator-activated receptor (PPAR)γ coactivator 1-alpha (SIRT1-PGC-1α)/NRF2 pathway, similar to the effects of aldose reductase inhibition in models of diabetic cardiomyopathy. Future studies testing this hypothesis in DCM models are warranted [27].
Energy metabolism derangements after DOX treatment were associated with a cardiac downregulation of PPAR-α [28], PPAR-γ [29], and PPAR-δ [30], which are transcription factors involved in the regulation of genes regulating carbohydrate, lipid, and protein metabolism. Administration of pharmacological agonists of PPARs was found to preserve heart function and reduce damage markers in murine models of DOX administration. Tumor-bearing wild-type mice receiving 4 mg/kg DOX every week for 6 weeks (to a final cumulative dose of 24 mg/kg) and fenofibrate (a PPAR-α agonist) display preserved heart function and cell survival compared to DOX-treated control group. This effect is recapitulated by overexpressing PPAR-α through a recombinant adeno-associated virus serotype 9 (rAAV9). The protection from DOX-associated toxicity exerted by PPAR-α activation is nullified by the mesenchyme homeobox 1 (MEOX1) knockdown, demonstrating a primary role for this factor in this model [28]. Mice receiving two weeks of pre-treatment with piperine, an agonist of PPAR-γ, and acute DOX treatment at 15 mg/kg display reduced cardiac dysfunction, ROS production, and cell death. These effects are lost if mice receive a pharmacological antagonist of PPAR-γ [29]. Similarly, Wistar rats receiving 15 mg/kg DOX and the PPAR-δ agonist GW0742 display preserved contractile function and reduced troponin phosphorylation compared to those receiving only DOX [30]. These results suggest that PPAR activation may be suitable for mitigating DOX-induced cardiotoxicity.
Mice receiving the lipid catabolism inducer adiponectin show reduced fibrosis and apoptosis after chronic DOX treatment, an effect lost if DOX is administered together with dorsomorphin, an AMP-activated protein kinase (AMPK) inhibitor [31]. AMPK is a keystone of energy metabolism in peripheral tissues. It responds to hormonal signals (e.g., adiponectin and leptin) to recover the energy status by stimulating glucose and lipid consumption and inhibiting anabolism [32]. In fact, knockdown of the glycolytic protein α-enolase rescues myocardial contraction, mitochondrial dysfunction, apoptosis, and AMPK dephosphorylation in the hearts of rats receiving chronic DOX administration [33]. AMPK activation was also observed in rats receiving oleuropein. This natural phenolic compound shows reduced metabolomic derangements after chronic DOX treatment, preserved cardiac function, reduced markers of inflammatory damage, and iNOS inhibition [34]. These results suggest that DOX induces detrimental cardiac effects by reducing AMPK activity.
Inflammatory balance is also crucial in DCM, as observed in mice receiving the interferon γ (IFNγ) inhibitor ‘R46-A2’ in acute DOX-induced cardiotoxicity that had improved cardiac function compared to those that received only DOX [35]. Of note, mice lacking the critical inflammatory receptor Toll-like receptor 4 (TLR-4) show preserved myocardial function, physiological inflammatory levels, and reduced apoptosis in response to acute DOX administration, demonstrating the relevance of inflammation in mediating the early cardiotoxicity of DOX [36]. Similarly, inflammasome-deficient Nod-like receptor protein 3 (NLRP-3) KO mice have reduced levels of DOX-induced pyroptosis and do not display myocardial structural and functional abnormalities after chronic DOX treatment. This mechanism involves the activation of NADPH oxidase 1 (NOX-1), NOX-4, and Caspase 1 by DOX, as proven by mechanistic experiments with Caspase 1 KO mice and pharmacological NOX inhibition (GKT137831), which abolish cardiotoxicity [37].
Overall, these studies show that preserving energy metabolism is a suitable strategy to mitigate the cardiotoxic effects of DOX (Figure 1).
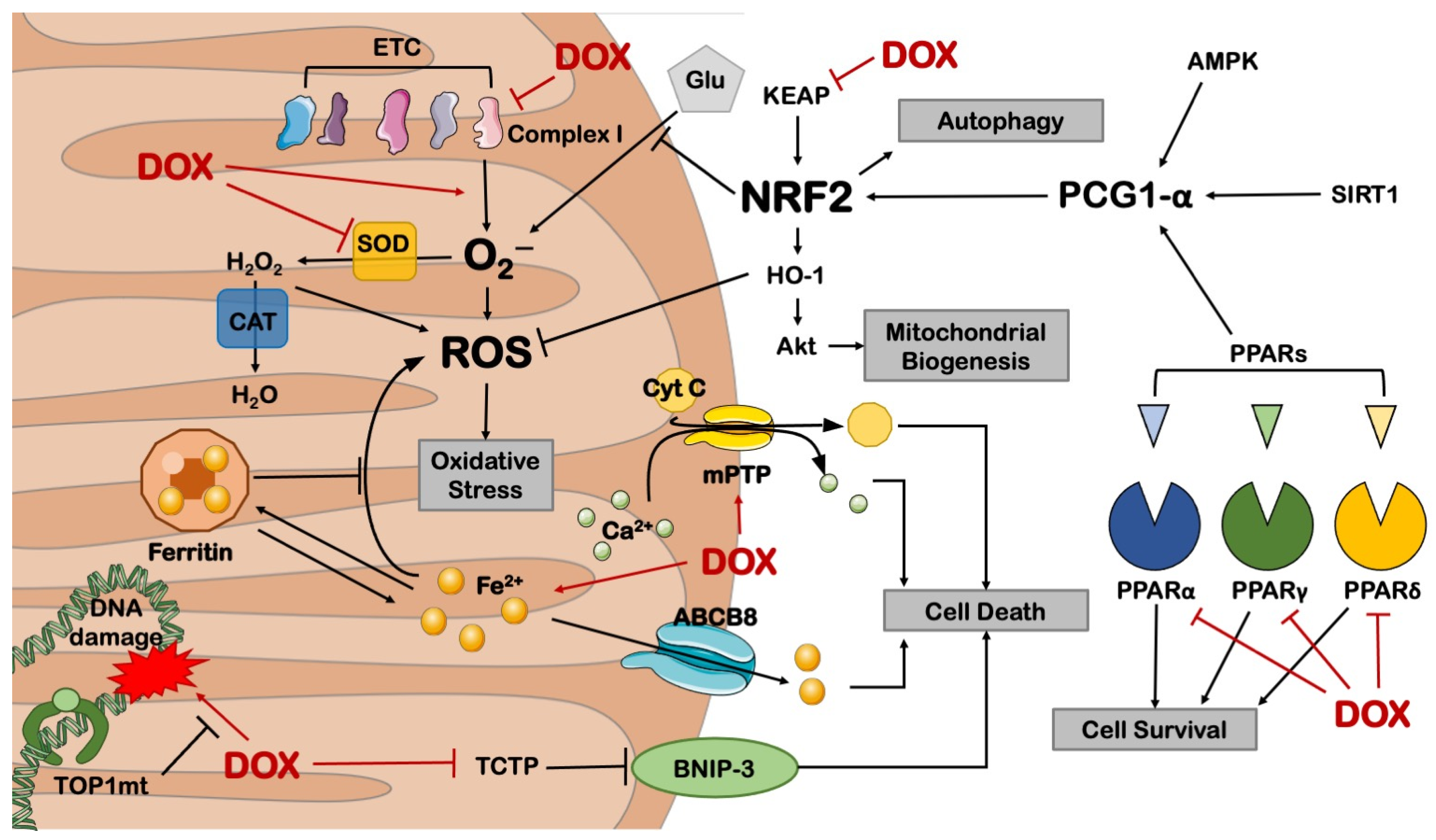
Figure 1. Mitochondrial derangements in DOX-induced cardiotoxicity. ABCB8 = ATP-binding cassette sub-family B member 8; AMPK = AMP-activated protein kinase; BNIP-3 = BCL2 19 kD protein-interacting protein 3; CAT = catalase; Cyt C = cytochrome C; ETC = electron transport chain; Glu = glucose; HO-1 = heme oxygenase 1; KEAP = Kelch-like ECH-associated protein; mPTP = mitochondrial permeability transition pore; NRF2 = nuclear respiratory factor 2; PGC-1α = PPARγ coactivator 1 alpha; PPAR = peroxisome proliferator-activated receptor; ROS = reactive oxygen species; SIRT1 = sirtuin 1; TCTP = translationally controlled tumor protein; TOP1mt = mitochondrial topoisomerase 1. This illustration includes elements from Servier Medical Art.
References
- Davies, K.J.; Doroshow, J.H. Redox cycling of anthracyclines by cardiac mitochondria. I. Anthracycline radical formation by NADH dehydrogenase. J. Biol. Chem. 1986, 261, 3060–3067.
- Doroshow, J.H.; Davies, K.J. Redox cycling of anthracyclines by cardiac mitochondria. II. Formation of superoxide anion, hydrogen peroxide, and hydroxyl radical. J. Biol. Chem. 1986, 261, 3068–3074.
- Goormaghtigh, E.; Chatelain, P.; Caspers, J.; Ruysschaert, J.M. Evidence of a specific complex between adriamycin and negatively-charged phospholipids. Biochim. Biophys. Acta 1980, 597, 1–14.
- Sinibaldi, F.; Howes, B.D.; Droghetti, E.; Polticelli, F.; Piro, M.C.; Di Pierro, D.; Fiorucci, L.; Coletta, M.; Smulevich, G.; Santucci, R. Role of lysines in cytochrome c-cardiolipin interaction. Biochemistry 2013, 52, 4578–4588.
- Wallace, K.B.; Sardão, V.A.; Oliveira, P.J. Mitochondrial Determinants of Doxorubicin-Induced Cardiomyopathy. Circ. Res. 2020, 126, 926–941.
- Aryal, B.; Rao, V.A. Deficiency in Cardiolipin Reduces Doxorubicin-Induced Oxidative Stress and Mitochondrial Damage in Human B-Lymphocytes. PLoS ONE 2016, 11, e0158376.
- Neilan, T.G.; Blake, S.L.; Ichinose, F.; Raher, M.J.; Buys, E.S.; Jassal, D.S.; Furutani, E.; Perez-Sanz, T.M.; Graveline, A.; Janssens, S.P.; et al. Disruption of nitric oxide synthase 3 protects against the cardiac injury, dysfunction, and mortality induced by doxorubicin. Circulation 2007, 116, 506–514.
- Huang, W.P.; Yin, W.H.; Chen, J.S.; Huang, P.H.; Chen, J.W.; Lin, S.J. Fenofibrate attenuates doxorubicin-induced cardiac dysfunction in mice via activating the eNOS/EPC pathway. Sci. Rep. 2021, 11, 1159.
- Ichikawa, Y.; Ghanefar, M.; Bayeva, M.; Wu, R.; Khechaduri, A.; Naga Prasad, S.V.; Mutharasan, R.K.; Naik, T.J.; Ardehali, H. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J. Clin. Investig. 2014, 124, 617–630.
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072.
- Fang, X.; Wang, H.; Han, D.; Xie, E.; Yang, X.; Wei, J.; Gu, S.; Gao, F.; Zhu, N.; Yin, X.; et al. Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. USA 2019, 116, 2672–2680.
- Zhang, H.; Wang, Z.; Liu, Z.; Du, K.; Lu, X. Protective Effects of Dexazoxane on Rat Ferroptosis in Doxorubicin-Induced Cardiomyopathy Through Regulating HMGB1. Front. Cardiovasc. Med. 2021, 8, 685434.
- Lipshultz, S.E.; Rifai, N.; Dalton, V.M.; Levy, D.E.; Silverman, L.B.; Lipsitz, S.R.; Colan, S.D.; Asselin, B.L.; Barr, R.D.; Clavell, L.A.; et al. The effect of dexrazoxane on myocardial injury in doxorubicin-treated children with acute lymphoblastic leukemia. N. Engl. J. Med. 2004, 351, 145–153.
- Haworth, R.A.; Hunter, D.R. The Ca2+-induced membrane transition in mitochondria. II. Nature of the Ca2+ trigger site. Arch. Biochem. Biophys. 1979, 195, 460–467.
- Montaigne, D.; Marechal, X.; Preau, S.; Baccouch, R.; Modine, T.; Fayad, G.; Lancel, S.; Neviere, R. Doxorubicin induces mitochondrial permeability transition and contractile dysfunction in the human myocardium. Mitochondrion 2011, 11, 22–26.
- Solem, L.E.; Heller, L.J.; Wallace, K.B. Dose-dependent increase in sensitivity to calcium-induced mitochondrial dysfunction and cardiomyocyte cell injury by doxorubicin. J. Mol. Cell. Cardiol. 1996, 28, 1023–1032.
- Marechal, X.; Montaigne, D.; Marciniak, C.; Marchetti, P.; Hassoun, S.M.; Beauvillain, J.C.; Lancel, S.; Neviere, R. Doxorubicin-induced cardiac dysfunction is attenuated by ciclosporin treatment in mice through improvements in mitochondrial bioenergetics. Clin. Sci. 2011, 121, 405–413.
- Baetz, D.; Regula, K.M.; Ens, K.; Shaw, J.; Kothari, S.; Yurkova, N.; Kirshenbaum, L.A. Nuclear factor-kappaB-mediated cell survival involves transcriptional silencing of the mitochondrial death gene BNIP3 in ventricular myocytes. Circulation 2005, 112, 3777–3785.
- Dhingra, R.; Guberman, M.; Rabinovich-Nikitin, I.; Gerstein, J.; Margulets, V.; Gang, H.; Madden, N.; Thliveris, J.; Kirshenbaum, L.A. Impaired NF-κB signalling underlies cyclophilin D-mediated mitochondrial permeability transition pore opening in doxorubicin cardiomyopathy. Cardiovasc. Res. 2020, 116, 1161–1174.
- Dhingra, R.; Margulets, V.; Chowdhury, S.R.; Thliveris, J.; Jassal, D.; Fernyhough, P.; Dorn, G.W., 2nd; Kirshenbaum, L.A. Bnip3 mediates doxorubicin-induced cardiac myocyte necrosis and mortality through changes in mitochondrial signaling. Proc. Natl. Acad. Sci. USA 2014, 111, E5537–E5544.
- Cai, W.; Fujita, T.; Hidaka, Y.; Jin, H.; Suita, K.; Shigeta, M.; Kiyonari, H.; Umemura, M.; Yokoyama, U.; Sadoshima, J.; et al. Translationally controlled tumor protein (TCTP) plays a pivotal role in cardiomyocyte survival through a Bnip3-dependent mechanism. Cell Death Dis. 2019, 10, 549.
- Yoshida, M.; Shiojima, I.; Ikeda, H.; Komuro, I. Chronic doxorubicin cardiotoxicity is mediated by oxidative DNA damage-ATM-p53-apoptosis pathway and attenuated by pitavastatin through the inhibition of Rac1 activity. J. Mol. Cell. Cardiol. 2009, 47, 698–705.
- Niu, Q.Y.; Li, Z.Y.; Du, G.H.; Qin, X.M. (1)H NMR based metabolomic profiling revealed doxorubicin-induced systematic alterations in a rat model. J. Pharm. Biomed. Anal. 2016, 118, 338–348.
- Yuan, Y.; Fan, S.; Shu, L.; Huang, W.; Xie, L.; Bi, C.; Yu, H.; Wang, Y.; Li, Y. Exploration the Mechanism of Doxorubicin-Induced Heart Failure in Rats by Integration of Proteomics and Metabolomics Data. Front. Pharmacol. 2020, 11, 600561.
- Sonowal, H.; Pal, P.B.; Wen, J.J.; Awasthi, S.; Ramana, K.V.; Srivastava, S.K. Aldose reductase inhibitor increases doxorubicin-sensitivity of colon cancer cells and decreases cardiotoxicity. Sci. Rep. 2017, 7, 3182.
- Sonowal, H.; Saxena, A.; Qiu, S.; Srivastava, S.; Ramana, K.V. Aldose reductase regulates doxorubicin-induced immune and inflammatory responses by activating mitochondrial biogenesis. Eur. J. Pharmacol. 2021, 895, 173884.
- Wang, Z.; Yuan, S.; Li, Y.; Zhang, Z.; Xiao, W.; Tang, D.; Ye, K.; Liu, Z.; Wang, C.; Zheng, Y.; et al. Regulation on SIRT1-PGC-1α/Nrf2 pathway together with selective inhibition of aldose reductase makes compound hr5F a potential agent for the treatment of diabetic complications. Biochem. Pharmacol. 2018, 150, 54–63.
- Wang, W.; Fang, Q.; Zhang, Z.; Wang, D.; Wu, L.; Wang, Y. PPARα Ameliorates Doxorubicin-Induced Cardiotoxicity by Reducing Mitochondria-Dependent Apoptosis via Regulating MEOX1. Front. Pharmacol. 2020, 11, 528267.
- Yan, J.; Xu, S.C.; Kong, C.Y.; Zhou, X.Y.; Bian, Z.Y.; Yan, L.; Tang, Q.Z. Piperine Alleviates Doxorubicin-Induced Cardiotoxicity via Activating PPAR-γ in Mice. PPAR Res. 2019, 2019, 2601408.
- Chen, Z.C.; Chen, L.J.; Cheng, J.T. Doxorubicin-Induced Cardiac Toxicity Is Mediated by Lowering of Peroxisome Proliferator-Activated Receptor δ Expression in Rats. PPAR Res. 2013, 2013, 456042.
- Konishi, M.; Haraguchi, G.; Ohigashi, H.; Ishihara, T.; Saito, K.; Nakano, Y.; Isobe, M. Adiponectin protects against doxorubicin-induced cardiomyopathy by anti-apoptotic effects through AMPK up-regulation. Cardiovasc. Res. 2011, 89, 309–319.
- Long, Y.C.; Zierath, J.R. AMP-activated protein kinase signaling in metabolic regulation. J. Clin. Investig. 2006, 116, 1776–1783.
- Gao, S.; Li, H.; Feng, X.J.; Li, M.; Liu, Z.P.; Cai, Y.; Lu, J.; Huang, X.Y.; Wang, J.J.; Li, Q.; et al. α-Enolase plays a catalytically independent role in doxorubicin-induced cardiomyocyte apoptosis and mitochondrial dysfunction. J. Mol. Cell. Cardiol. 2015, 79, 92–103.
- Andreadou, I.; Mikros, E.; Ioannidis, K.; Sigala, F.; Naka, K.; Kostidis, S.; Farmakis, D.; Tenta, R.; Kavantzas, N.; Bibli, S.I.; et al. Oleuropein prevents doxorubicin-induced cardiomyopathy interfering with signaling molecules and cardiomyocyte metabolism. J. Mol. Cell. Cardiol. 2014, 69, 4–16.
- Ma, P.; Qin, Y.; Cao, H.; Erben, U.; Ni, C.; Qin, Z. Temporary blockade of interferon-γ ameliorates doxorubicin-induced cardiotoxicity without influencing the anti-tumor effect. Biomed. Pharmacother. 2020, 130, 110587.
- Riad, A.; Bien, S.; Gratz, M.; Escher, F.; Westermann, D.; Heimesaat, M.M.; Bereswill, S.; Krieg, T.; Felix, S.B.; Schultheiss, H.P.; et al. Toll-like receptor-4 deficiency attenuates doxorubicin-induced cardiomyopathy in mice. Eur. J. Heart Fail. 2008, 10, 233–243.
- Zeng, C.; Duan, F.; Hu, J.; Luo, B.; Huang, B.; Lou, X.; Sun, X.; Li, H.; Zhang, X.; Yin, S.; et al. NLRP3 inflammasome-mediated pyroptosis contributes to the pathogenesis of non-ischemic dilated cardiomyopathy. Redox Biol. 2020, 34, 101523.
More