Glucagon-like peptide-1 (GLP-1) is a human incretin hormone derived from the proglucagon molecule. GLP-1 receptor agonists are frequently used to treat type 2 diabetes mellitus and obesity. However, the hormone affects the liver, pancreas, brain, fat cells, heart, and gastrointestinal tract. The results showed that GLP-1 agonists can benefit defined off-medication motor scores in Parkinson’s Disease and improve emotional well-being. In Alzheimer’s disease, GLP-1 analogs can improve the brain’s glucose metabolism by improving glucose transport across the blood–brain barrier. In depression, the analogs can improve quality of life and depression scales. GLP-1 analogs can also have a role in treating chemical dependency, inhibiting dopaminergic release in the brain’s reward centers, decreasing withdrawal effects and relapses. These medications can also improve lipotoxicity by reducing visceral adiposity and decreasing liver fat deposition, reducing insulin resistance and the development of non-alcoholic fatty liver diseases. The adverse effects are primarily gastrointestinal. Therefore, GLP-1 analogs can benefit other conditions besides traditional diabetes and obesity uses.
1. Introduction
Glucagon-like peptide-1 (GLP-1) is an incretin secreted by the distal intestinal ileum and colon L-cells following food intake. The synthesis of this molecule occurs through the proteolytic cleavage of proglucagon molecules performed by many prohormone convertase enzymes. The cleavage of proglucagon leads to the creation of two active peptides 30 or 31 amino acids long: GLP-1(7-36)-NH2 (the most commonly produced molecule) and GLP-1 (7-37) (the second most commonly produced molecule)
[1].
GLP-1 has a short half-life (around 2 min), due to the action of the enzyme dipeptidyl-peptidase-4 (DPP-4), a serine aminopeptidase, and because of renal clearance, due to its low molecular weight. The DPP-4 endopeptidase is present in the membrane of cells of multiple organs (kidney, liver, pancreas, blood vessels, gut, and brain) and in a soluble form
[1][2][1,2].
The biological effects of GLP-1 are mediated via binding to G protein-coupled transmembrane receptors (GPCRs) of the class B family. After hormone–receptor ligation, the levels of cAMP increase by the increased activity of adenylate cyclase, and the different actions of GLP-1 occur
[1][2][1,2].
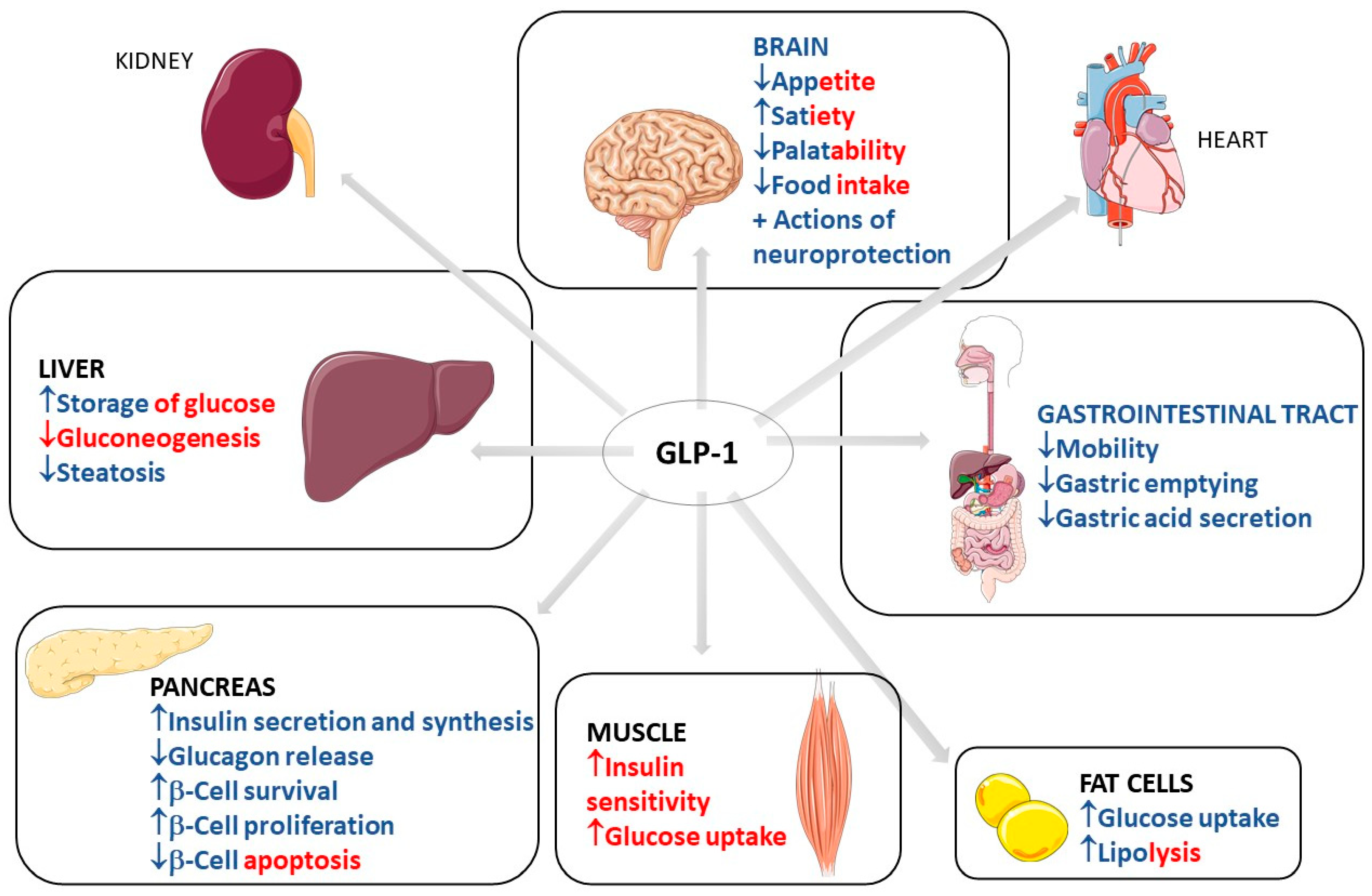
GLP-1 has biological effects that are well known and well established in clinical medicine. These effects occur through the binding to GLP-1 receptors in multiple organs and body systems. GLP-1 action in the pancreas includes an increase in insulin secretion and synthesis, an increase in pancreatic β-cell proliferation and β-cell survival, and a decrease in glucagon release. This scenario leads to reduced gluconeogenesis and increased hepatic storage of glucose as glycogen. Other actions include increased glucose uptake by muscle cells and increased glucose uptake and increased lipolysis in adipocytes. GLP-1 can also reduce appetite, gastrointestinal motility, and gastric acid secretion. The actions of GLP-1 in the kidney include mild natriuresis. GLP-1 increases contractility and heart rate in the heart and has vascular protective effects. Due to these metabolic effects of GLP-1, it is primarily used in clinical practice to treat type 2 diabetes mellitus (T2DM) and obesity [1,2,3,4,5,6,7]. shows the most important traditional organ targets for GLP-1 activities and its actions on each target, with direct and indirect effects differentiated by color.
Figure 1. Most important traditional organ targets for GLP-1 and its actions on each target. GLP-1: Glucagon-like peptide; ↓: decrease; ↑: increase; +: plus. The red color represents the indirect effects of GLP-1 on the determined organ, and the blue color represents direct effects. The red–blue mixtures represent the determining effects in direct and indirect related GLP-1 activity.
2. Use of GLP1
2.1. Common Use of GLP-1: Diabetes and Obesity
The relation between obesity and DM is already well explained, and it is known that diabetes is closely related to increased cardiovascular risk. Obesity causes health problems proportionally with the increase of the individual’s body mass index (BMI), leading to increased mortality and morbidity, mainly in cases with a BMI > 30 kg/m
2. In addition, obesity also can lead to many other outcomes, such as hypertension and abnormal lipid metabolism that leads to dyslipidemia, endothelial dysfunction, and augmented proinflammatory state
[8][9][37,38].
The insulin resistance environment is produced when an abnormal ectopic deposition of fat is observed mainly in the skeletal muscles and the liver. Furthermore, genetic factors and phenotypical interactions can increase metabolic diabetes risk and contribute to the residual β-cells that cannot adequately control insulin secretion in the right proportions to overcome the insulin resistance state
[8][10][11][37,39,40].
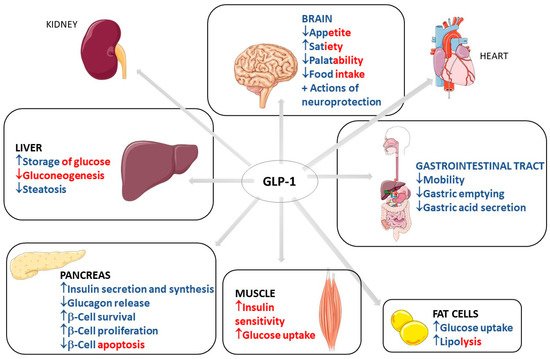
The usual actions of GLP-1 are summarized as controlling energy homeostasis and feeding behavior. GLP-1 regulates energy homeostasis principally by its effects on stimulating insulin secretion to control glycemic excursions and its actions on inhibiting glucagon secretion. Moreover, GLP-1 controls feeding behavior by affecting multiple neural circuits that correspond to appetite. In addition, it is known that GLP-1 affects gastric emptying and inhibits pancreatic β-cells apoptosis.
The short-acting (2–5 h half-life) and the long-acting (several to 12 h half-life) GLP-1 receptor agonists have considerable effects on the treatment of not only diabetes but also obesity. The effectiveness of these drugs on diabetes and obesity is primarily due to the regulation of glycemia, insulin, and glucagon secretion. Short-acting GLP-1 receptor agonists have demonstrated modest reductions of fasting blood glucose levels and substantial reductions of postprandial hyperglycemia by stimulating fasting insulin secretions and decreasing glucagon secretion. In addition, these molecules reduce the gastric emptying rate and reduce appetite, leading to a reduction of the body weight by 1–5 kg on average. On the other hand, long-acting GLP-1 agonists have shown substantial decreases in fasting blood glucose levels and modest reductions of postprandial hyperglycemia with strong stimulations of fasting insulin secretions and reductions of glucagon secretion. As a result, the long-acting GLP-1 agonists may cause a decrease of body weight by 2–5 kg on average. It is known that the actions of the native human GLP-1 are different in reducing food intake and in controlling blood glucose levels in comparison to GLP-1 receptor agonists. Recent findings demonstrated that in overweight or obese adults, GLP-1 receptor agonists and phentermine–topiramate are the best drugs to reduce body weight; of the GLP-1 agonists, semaglutide might be the most effective
[12][13][14][15][16][43,44,45,46,47].
Figure 2 summarizes the GLP-1 actions.
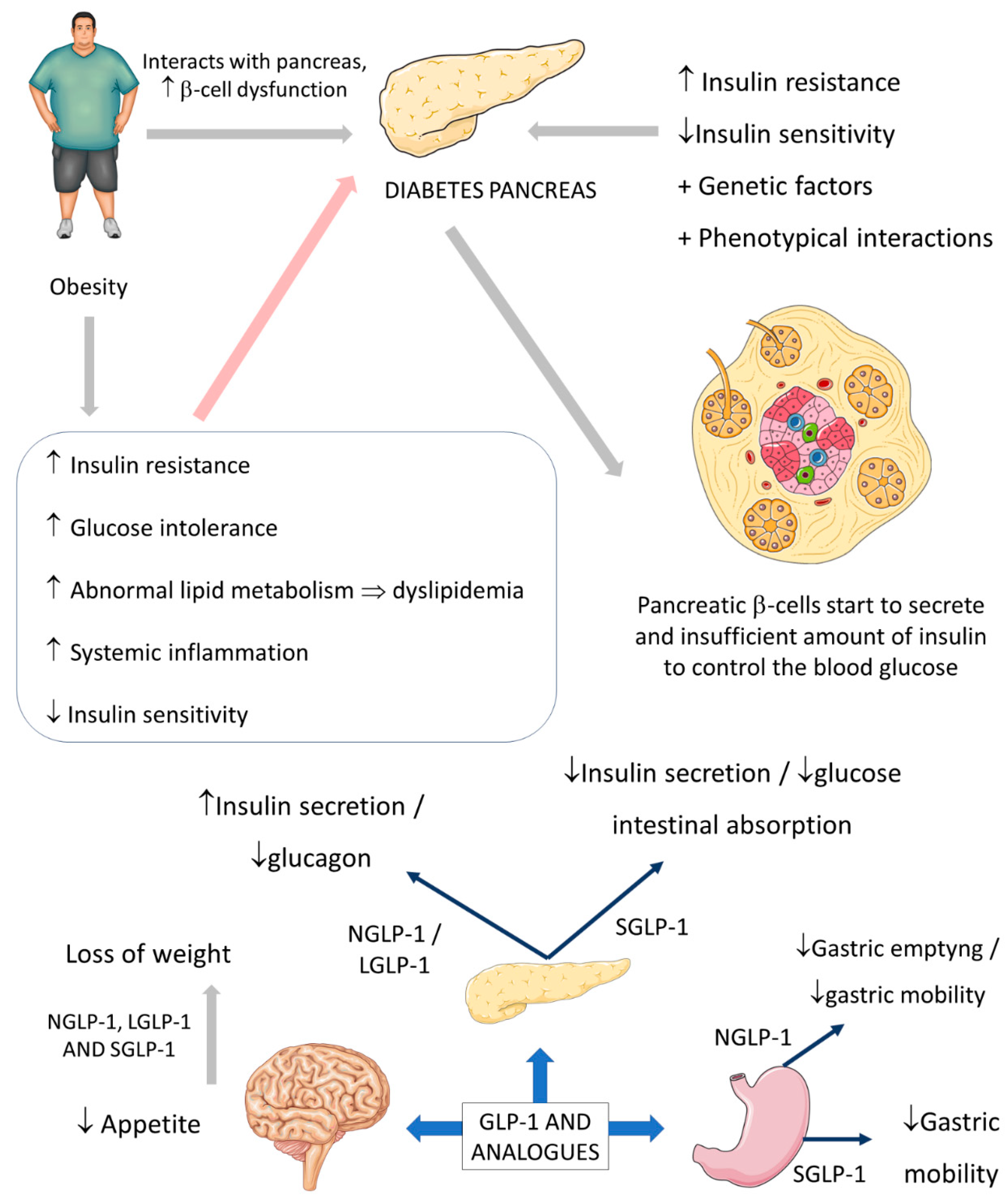
Figure 2. Diabetes, obesity, and possible actions of GLP-1. GLP-1: glucagon-like peptide; NGLP-1: native GLP-1; LGLP-1: long-acting GLP-1 receptor agonist; SGLP-1: short-acting GLP-1 receptor agonist; ↓: decrease; ↑: increase; ⇒: equal; +: plus.
2.2. GLP-1 and Parkinson’s Disease
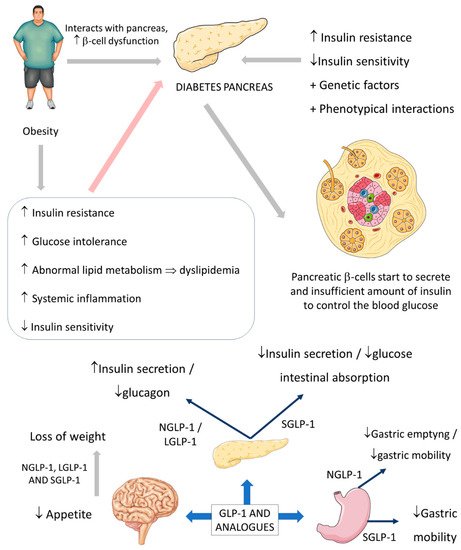
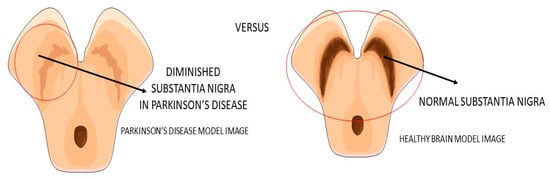
Parkinson’s disease (PA) is a progressive neurodegenerative disease (
Figure 3) characterized predominantly by motor signals, such as tremor, akinesia, and bradykinesia. However, nonmotor symptoms and signs can also be found, such as cognitive decline, depression, signals of anxiety, sleep disturbances, and dysautonomia. It is a common neurologic ailment that affects 1.5% of individuals over 65 years old worldwide. In Parkinson’s, midbrain neurons are degenrated mainly because of mitochondrial dysfunctions that lead to cellular energy failure and, consequently, to a reduction of dopamine transmission and deposition of α-synuclein, in addition to the presence of increased oxidative stress. The deposition of a-synuclein, accumulation of reactive oxygen species (ROS), and the presence of dying neuron cells trigger an inflammatory environment in the midbrain tissue. The process leads to degeneration of the brain, together with autophagy and apoptosis. The gold standard treatments are dopamine replacement with dopaminergic medications or brain stimulation. These treatments improve the motor signals, but they are only partially effective due to the progressive nature of Parkinson’s. Subsequently, the non-dopaminergic systems will be involved in the progression of the disease, causing some nonmotor symptoms, for instance, depression and cognitive decline
[17][18][19][49,50,51].
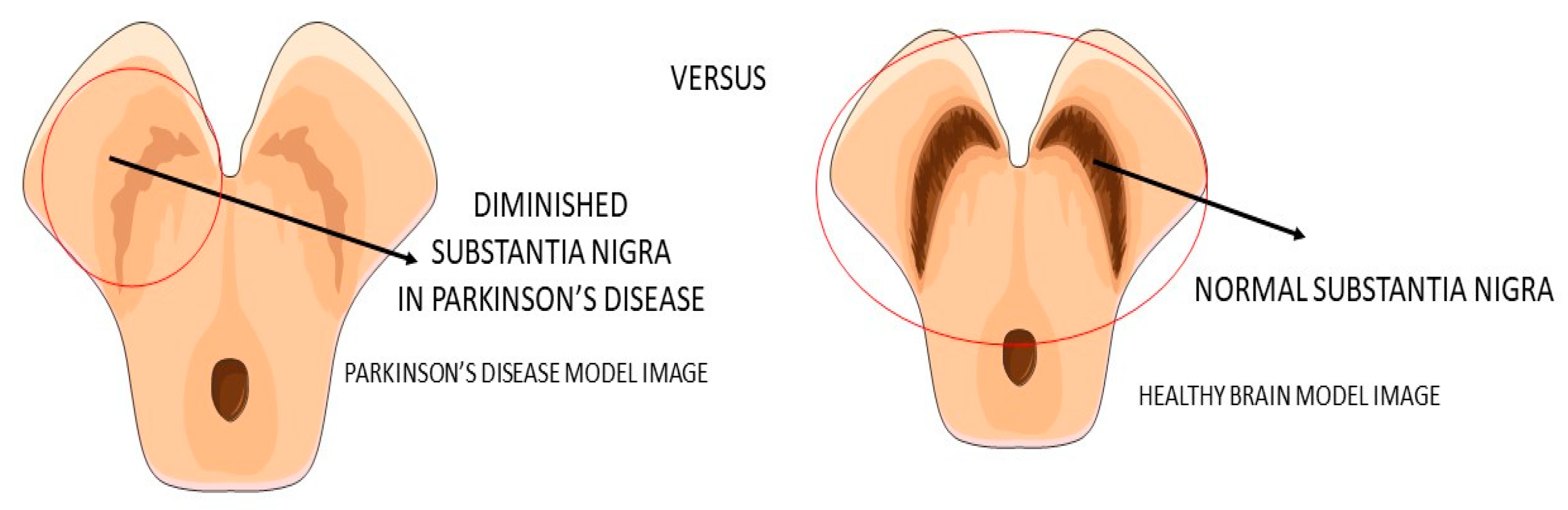
Figure 3. Substantia nigra in Parkinson’s disease and the healthy brain.
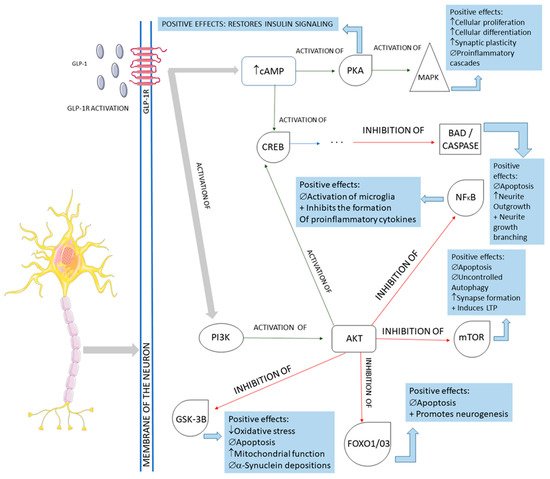
The beneficial actions of GLP-1 go far beyond insulin secretion and appetite and include cardiovascular benefits and possibly also beneficial effects in neurodegenerative diseases. GLP-1 presents neurotrophic and neuroprotective effects demonstrated by some clinical trials studies and mouse model studies. It can cross the blood–brain barrier and central nervous system and may influence some neural actions in cellular pathways, such as neuroinflammation, mitochondrial dysfunction, and cellular proliferation. For example, the decontrolled mitochondrial functions may lead to cellular energy failure. This procedure is implicated as the leading cause of dopaminergic neuron death in Parkinson’s disease and other age-related neural dysfunction.
The activation of the neurons of the central nervous system by GLP-1 and the interferences of this molecule in the pathological pathways of Parkinson’s disease are shown in
Figure 4. In summary, GLP-1 molecules will bind to the GLP-1Rs located on the membrane of the midbrain’s dopaminergic neurons and activate these cells against the typical pathological pathways of Parkinson’s disease. In Parkinson’s, these midbrain neurons are degenerated mainly because of mitochondrial dysfunctions that lead to a reduction of dopamine transmission and the deposition of a-synuclein, in addition to the presence of increased oxidative stress. The deposition of a-synuclein, the accumulation of reactive oxygen species (ROS), and the presence of dying neuron cells trigger an inflammatory environment in the midbrain tissue. The GLP-1 binding on the GLP-1Rs starts several signalizing pathways in the neurons, reducing inflammation, oxidative stress, apoptosis, and a-synuclein deposition. In addition, cell proliferation restores insulin signaling and improves neuronal functions. Restoring insulin signaling in these neurons corresponds to a neuroprotective action of GLP-1 in diabetic individual neurodegeneration since T2DM subjects present insulin resistance of the brain cells that may accelerate the progression of Parkinson’s disease
[17][20][21][22][23][24][25][26][27][9,21,49,52,54,55,56,57,58].
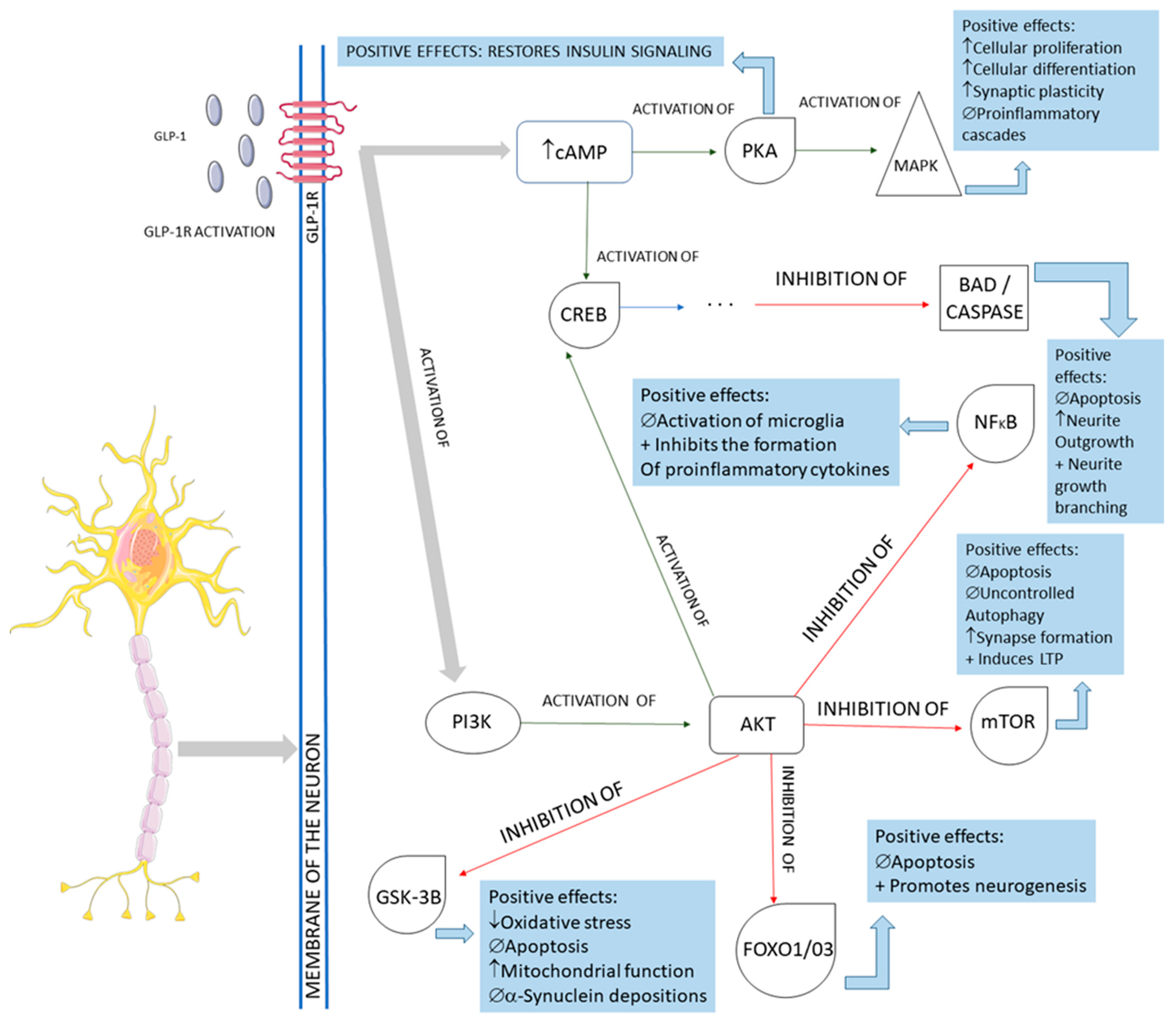
Figure 4. Main cellular pathways affected by GLP-1 in neurons. GLP-1R: GLP-1 receptor; cAMP: cyclic adenosine monophosphate; PKA: protein kinase A; MAPK: mitogen-associated protein kinase; CREB: cyclic adenosine monophosphate response element-binding protein; BAD: Bcl-2 antagonist of death; NFkB: nuclear factor-kappa B; PI3K: phosphoinositide 3-kinase; AKT: protein kinase B; mTOR: mammalian target of rapamycin; GSK-3B: glycogen synthase kinase 3 beta; FOXO1/03: forkhead box protein O1; ↓: decrease; ↑: increase; ∅ = impairment; +: plus.
2.3. GLP-1, Non-Alcoholic Fatty Liver Disease, and Non-Alcoholic Steatohepatitis
3.3. GLP-1, Non-Alcoholic Fatty Liver Disease, and Non-Alcoholic Steatohepatitis
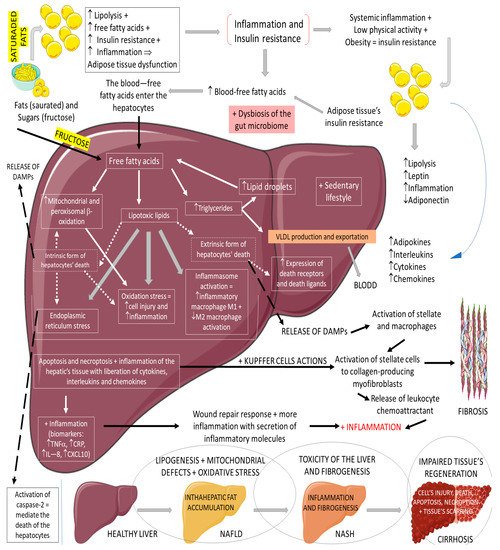
Non-alcoholic fatty liver disease (NAFLD) is the most common cause of chronic liver disease in western countries. It can be characterized by an excessive deposition and accumulation of fatty molecules in the hepatocytes in the absence of secondary causes. This accumulation can be an isolated event or accompanied by liver inflammation, which causes non-alcoholic steatohepatitis (NASH). Hepatic tissue inflammation causes progressive cell injury on the hepatocytes that can lead to fibrosis. In a variable course, the progression of the disease can lead, in addition to cirrhosis, to hepatocellular carcinoma. It is known that the massive presence of fatty molecules in the liver’s tissue is highly associated with risk factors for metabolic syndrome (MS), obesity, and insulin resistance. Although NAFLD and NASH share similar pathological pathways with alcohol-related fatty liver disease (ALD), NAFLD and NASH are considered metabolic disorders mainly characterized by the accumulation of triglycerides in hepatocytes. Nowadays, the prevalence of NAFLD and NASH is rising primarily due to the increased incidence of diabetes and MS. It is known that MS corresponds to a very high risk factor for NAFLD and NASH. MS is characterized by central abdominal obesity, systemic hypertension, insulin resistance (or T2DM), and atherogenic dyslipidemia, contributing to a prothrombotic and proinflammatory state. This scenario leads to an increase in oxidative stress (OS), which is related to mitochondrial dysfunction, accumulations of protein and oxidated lipids, and an impairment of the antioxidant system
[28][29][30][31][32][33][34][64,65,66,67,68,69,70]. The impaired lipid metabolism in NAFLD and NASH is worsened by the increase of free fatty acid influxes to the hepatocytes and hepatic insulin resistance, in addition to a sedentary lifestyle
[28][29][30][31][32][33][34][64,65,66,67,68,69,70].
Figure 5 summarizes the physiopathology of NAFLD and the progression to NASH.
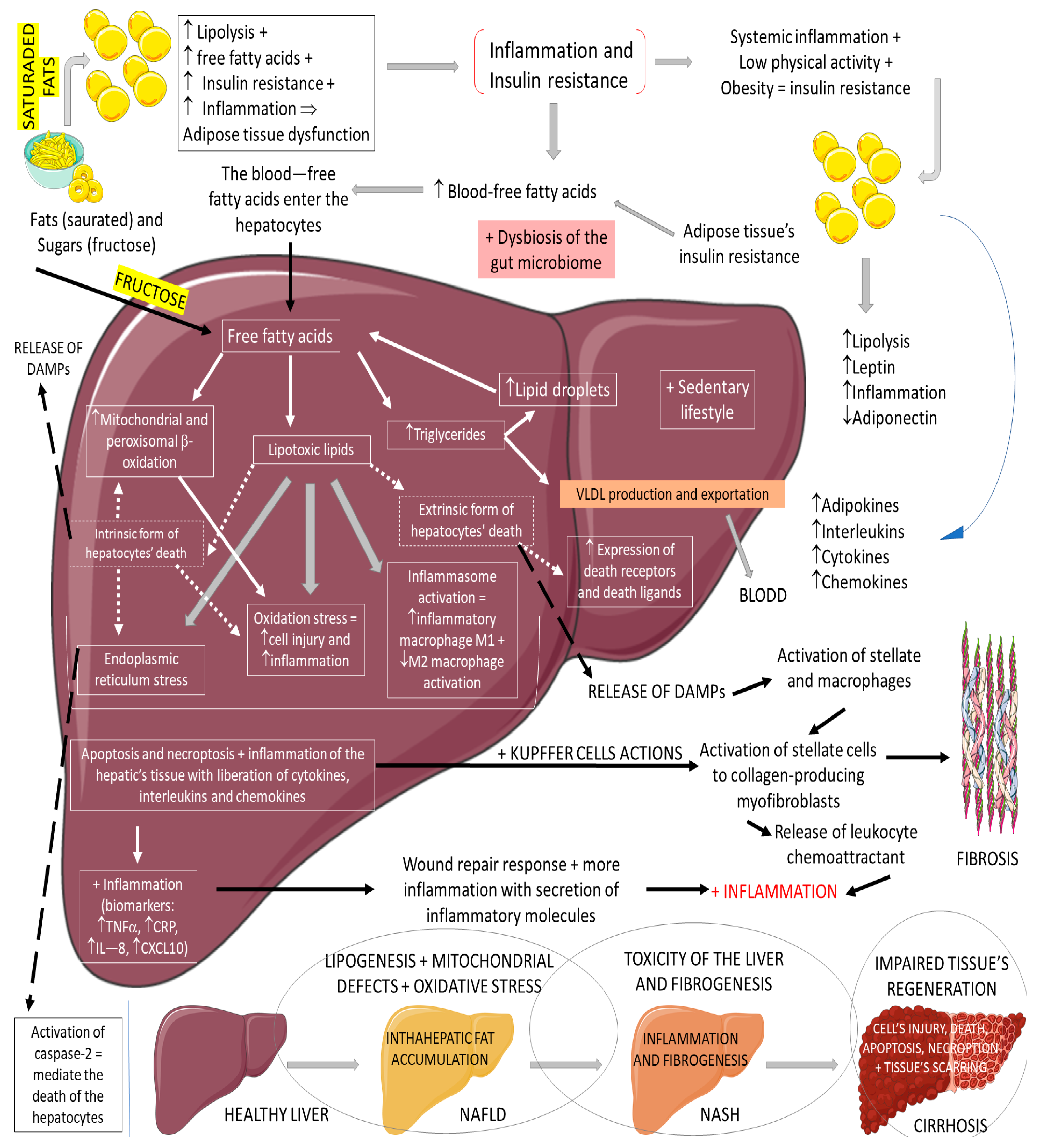
Figure 5. Physiopathology of NAFLD and the progression to NASH. ↓: decrease; ↑: increase; +: added to; ⇒: equal; TNF-α: tumor factor necrosis; CRP: C reactive protein; IL-8: interleukin 8; CXCL10: C–V–C motif chemokine ligand 10.
2.4. GLP-1 and Alzheimer’s Disease
3.4. GLP-1 and Alzheimer’s Disease
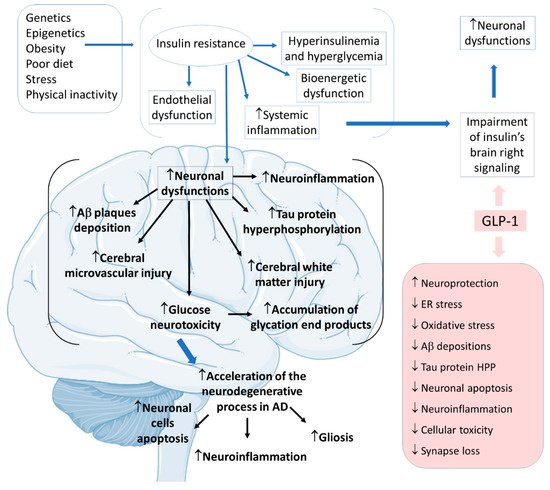
Alzheimer’s disease (AD) is the most common neurodegenerative cause of dementia worldwide, affecting more than 10% of people over 65. Metabolic, mitochondrial, inflammatory, oxidative, and vascular changes are associated with the development of AD. However, the most significant hypothesis for the pathophysiology derives from the amyloid cascade. The amyloid precursor protein (APP) is a type I transmembrane protein synthesized primarily in the endoplasmic reticulum and posteriorly transported to the Golgi apparatus to be stored at a steady state in its final synthesis process. Subsequently, the APP molecules can be processed and cleaved at different sites by the actions of other proteases. In this protein processing, changes in amyloid cleavage can lead to the formation of an APP fragment called β-amyloid (Ab). The presence of these Ab molecules can make Ab encounter hyperphosphorylated tau protein aggregations. Consequently, this coalescence leads to a reduction in synaptic strength, loss of synaptic connections, and neurodegeneration. In addition to this interaction of proteins, it is known that AD is closely banded with peripheral and brain insulin resistance, insofar as the impaired insulin signaling in the brain contributes to amplifying neuronal dysfunctions and neurodegeneration
[35][36][37][38][39][75,76,77,78,79].
GLP-1 is also produced in the central nervous system of humans, predominantly in the brainstem neuronal cells of the nucleus tractus solitarius of the central nervous system, to be distributed to other central nervous areas, such as the hypothalamus thalamic and cortical areas. GLP-1 triggers in vivo actions by binding to G proteins to activate cellular pathways that lead to various effects in the central nervous areas. Not only the insulin resistance of the neurons but systemic hyperinsulinemia corresponds to the harmful impact on cognitive impairment
[2][35][36][37][38][39][40][41][42][2,75,76,77,78,79,80,81,82].
Considering insulin resistance as a critical part of the AD pathogenic pathway, GLP-1 and its analogs can have beneficial actions on treating neurodegenerative states. GLP-1 has central effects on neuroprotection, which can be summarized by increases in neurogenesis and synaptic plasticity and decreases in neuroinflammation and protein aggregations. The pathological cascade of brain insulin resistance lies in the fact that this resistance increases Aβ plaque deposition and tau protein hyperphosphorylation in the interior of the affected AD brain areas. For these reasons, it is believed that in AD, GLP-1 reduces neuronal apoptosis, neuroinflammation, and the typical gliosis. GLP-1 can also show beneficial effects in reducing endoplasmic reticulum stress, oxidative stress, Aβ depositions, tau protein hyperphosphorylation, cellular toxicity, and synaptic loss. Furthermore, GLP-1 can amplify insulin signaling in the brain cells, leading to increased insulin sensitivity in the neurons. In addition, the insulin resistance that strengthens Aβ depositions and tau protein hyperphosphorylation can cause microvascular injury and white matter injury in the brain. Moreover, glucose neurotoxicity and accumulation of advanced glycation end products in the brain is observed. The treatment with GLP-1 can effectively prevent these events (
Figure 6)
[2][35][36][37][38][39][40][41][42][2,75,76,77,78,79,80,81,82].
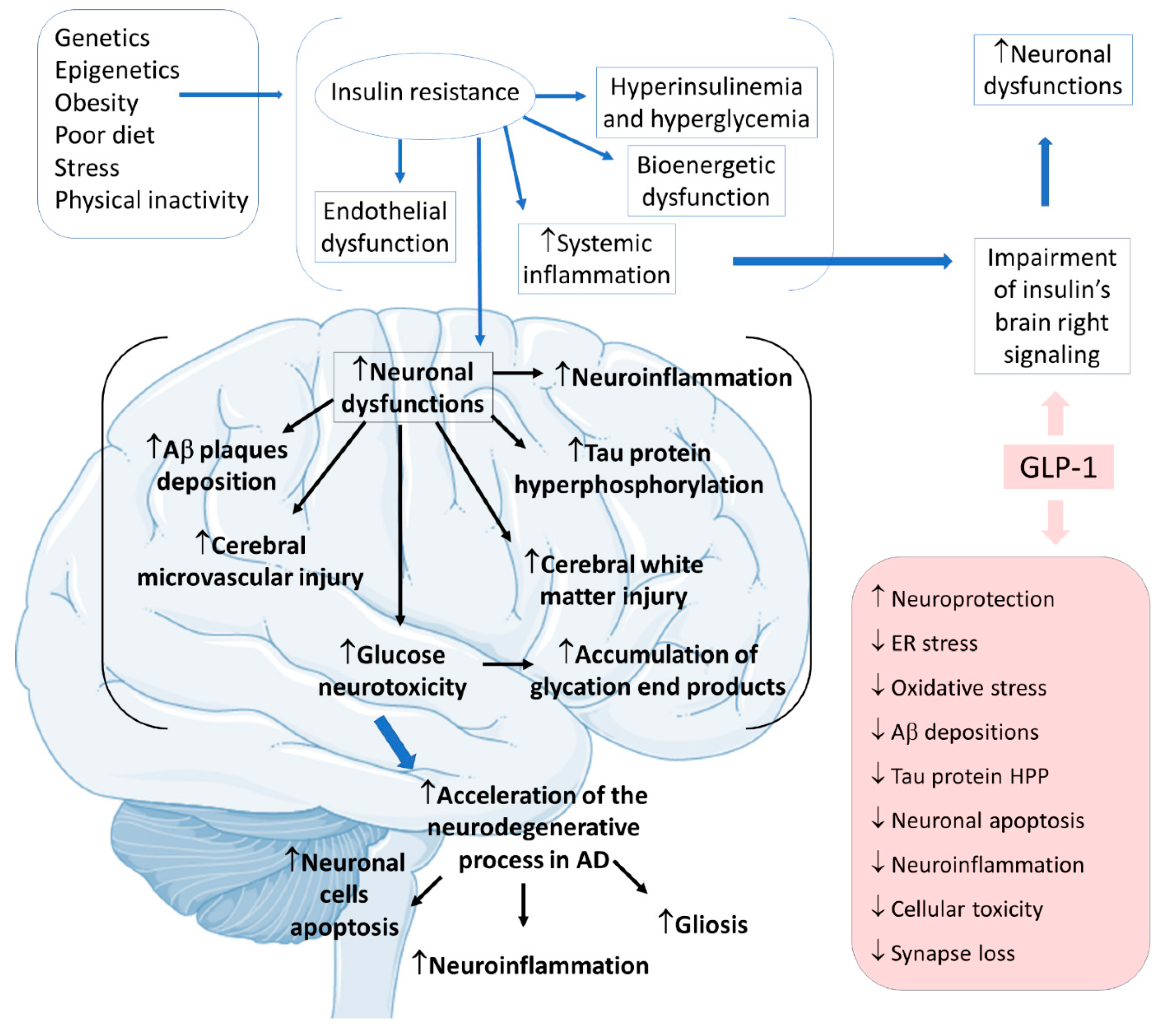
Figure 6. Pathophysiology of Alzheimer’s disease focused on insulin resistance and the effects of GLP-1. Ad: Alzheimer’s disease; ↓: decrease; ↑: increase; Aβ: β-amyloid; ER: endoplasmic reticulum; HPP: hyperphosphorylation.
2.5. GLP-1 and Depression
3.5. GLP-1 and Depression
Depression is a common debilitating mental condition associated with a profound impact on the quality of life, mainly due to poor management related to under-diagnosis and under-treatment. Depression is also emerging in the globalized world as one of the most important causes of morbidity and mortality. Although depression is a heterogeneous syndrome that comprehends complex pathogenesis, some neurobiological factors influence this condition. In addition, anatomical modifications in the prefrontal cortex, amygdala, hippocampus, and all areas of the central nervous system can contribute to the development of this condition. Furthermore, it is known nowadays that genetic and environmental factors are also involved in the pathophysiology of depression. However, despite all the research in this area, this condition is still not fully understood. Recent studies have shown the relationship between depression and metabolism, since diabetes and obesity were already considered conditions related to a significant risk factor for neurophysiological conditions. In summary, depressive diseases correspond to somatic and cognitive alterations principally including sadness, empty moods, irritability, or anhedonia. It is known that if patients affected by depression do not receive adequate treatment, the depressive symptoms aggravate and serious outcomes can emerge
[43][44][45][46][47][83,84,85,86,87].
Neuroinflammation is one condition intimately related to depression due to sustained stress. The peripheral levels of inflammatory markers can correspond to the severity of depression. Patients with depressive disorders exhibit high levels of proinflammatory cytokines, such as IL-1β, TNF-α, IL-6, and IL-1, followed by CRP elevation. In addition, neuroinflammation is associated with an increase in OS in the brain. The brain presents high amounts of polyunsaturated fatty acids in its composition, inadequate anti-oxidative machinery, and consumes large amounts of oxygen. Chronic neuroinflammation is related to serotonergic, dopaminergic, and noradrenergic dysfunctions, and the modulation of inflammatory conditions can be associated with ameliorations in depressive symptoms. GLP-1 can potentially protect against neuroinflammation principally by controlling the production of proinflammatory cytokines and reducing OS
[44][45][48][84,85,88].