The endoplasmic reticulum (ER) is site of synthesis and maturation of membrane and secretory proteins in eukaryotic cells. The ER contains more than 20 members of the Protein Disulfide Isomerase (PDI) family. These enzymes regulate formation, isomerization and disassembly of covalent bonds between cysteine residues. As such, PDIs ensure protein folding, which is required to attain functional and transport-competent structure, and protein unfolding, which facilitates dislocation of defective gene products across the ER membrane for ER-associated degradation (ERAD). The PDI family includes over a dozen of soluble members and few membrane-bound ones. Among these latter, there are five PDIs grouped in the thioredoxin-related transmembrane (TMX) protein family.
- endoplasmic reticulum
- protein folding
- ERAD
- PDI
- TMXs
1. Introduction
About one third of the proteome in eukaryotic cells is made of membrane and secretory proteins [1]. Their production and maturation occurs within the ER with help and under surveillance of resident chaperones and folding enzymes, such as the members of the PDI family [2]. PDIs assist protein folding by catalyzing the formation of the native set of intra- and inter-molecular disulfide bonds (oxidation); they can also correct structural errors by disassembling non-native disulfides to promote their conversion into the native set (isomerization) [3][4]; they can facilitate the translocation across the ER membrane of terminally misfolded polypeptides by dissolving intra- and inter-molecular disulfide bonds (reduction), in a step that precedes their degradation by cytosolic 26S-proteasomes [5][6]. In addition to these activities, PDIs can also act as regulators of the luminal calcium homeostasis [7] and participate to multimeric structures such as the prolyl 4-hydroxylase [8] or the oligosaccharyltransferase complexes [9].
More than 20 PDI family members have been identified, so far [10]. The reasons for such a high number is not fully understood. However, their tissue distribution, membrane topology, and organization of the active site hint at client-specificity and high functional versatility [11]. Most PDI family members are soluble in the ER lumen, with few membrane-anchored exceptions [4]. The TMX protein family comprises five membrane-tethered PDIs (TMX1, TMX2, TMX3, TMX4 and TMX5) [12][13][14][15][16] (Figure 1 and Table 1). These proteins are all characterized by an N-terminal signal sequence for ER targeting and one catalytically active thioredoxin (TRX)-like domain (known as type-a TRX-like domain), containing the active site. TMX1, the best characterized member of the TMX family, preferentially interacts with membrane-bound folding-competent and folding-defective clients [17][18]. In contrast, the other members of the family have been poorly studied, if at all.
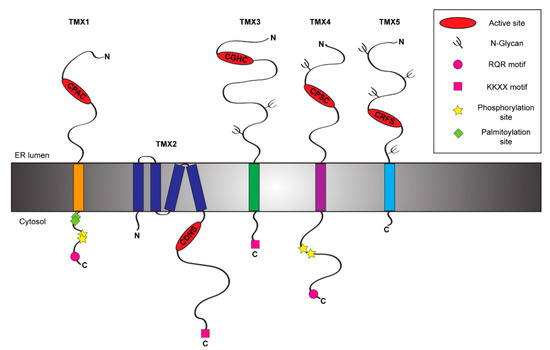
Figure 1. Schematic representation of the TMX protein family members. The figure shows the topology and the main structural and functional features of the five members of the TMX family [12][13][14][15][16].
Table 1. List of the TMX family members. The table displays the main features of the five TMXs including their active site sequences and biological functions. a, active type-a TRX-like domain; b, inactive type-b TRX-like domain; R, reductase activity; O, oxidase activity.
|
Protein |
TRX-like domains |
Active site |
Activities |
|
Biological functions |
|
TMX1 |
a |
CPAC |
R |
|
Protein folding and ERAD Ca2+ flux regulation |
|
TMX2 |
a |
SNDC |
? |
|
Nuclear protein import Ca2+ flux regulation |
|
TMX3 |
abb' |
CGHC |
O |
|
? |
|
TMX4 |
a |
CPSC |
R |
|
Protein folding |
|
TMX5 |
a |
CRFS |
? |
|
? |
2. TMX1: A Topology-Specific ER-Resident Reductase
TMX1 (other name TXNDC1) is the best-known member of the TMX family. It has been identified in 2001 by Matsuo and colleagues [16] among the genes up-regulated by TGF-β [19]. TMX1 is a single-pass type I protein of 280 residues with a large luminal N-terminal region harboring a TRX-like domain and a short cytosolic tail [16] (Figure 1). TMX1 displays a di-arginine motif that ensures its retention within the ER [16][20][21] (Figure 1). The cytosolic tail of TMX1 also contains both palmitoylation [22] and phosphorylation sites [23] (Figure 1). These modifications affect the sub-ER localization of TMX1 and may determine the spectrum of clients [21]. TMX1 is ubiquitously expressed in human tissues, with the highest levels in kidney, lungs, placenta and liver [16]. Unlike other members of the PDI family, TMX1 does not contain an ER stress responsive element (ERSE) within its promoter region [24] and indeed it is not up-regulated upon ER stress [25]. Deletion of the TMX1 gene is innocuous at the cellular level. This suggests the activation of compensatory mechanisms in cultured cells, where other members of the PDI family may play a role [26]. At the organism level, however, the absence of TMX1 has consequences as it increases susceptibility to liver damage in mice challenged with lipopolysaccharides [26]. From a functional point of view, TMX1 displays a non-canonical CPAC active site in its type-a TRX-like domain [16] (Figure 1 and Table 1). The proline in position 2 suggests a role as reductase [27], since it destabilizes the disulfide state and favors the di-thiol reduced form of the active site [28]. Consistently, TMX1 is predominantly reduced in vivo [25], and in vitro it reduces insulin disulfides [16]. Of additional support to the putative function of TMX1 as an ER reductase, it has been demonstrated that TMX1 overexpression enhances the cytotoxicity of the toxins ricin and abrin, two type 2 ribosome-inactivating proteins requiring a step of reduction in the ER before the dislocation of the catalytic subunits to the cytosol [29]. TMX1 has been also found to interact with vitamin K epoxide reductase (VKOR), an enzyme involved in the process of blood coagulation working with membrane-tethered TRX-like proteins, which serve as redox partners [30].
From the functional point of view, TMX1 represents the first example of topology-specific redox catalyst involved in both protein folding and ERAD pathways [17][18]. Indeed, TMX1 recruits membrane-bound N-glycosylated clients through a cooperative interaction with the ER lectin CNX catalyzing their folding [17]. Moreover, TMX1 reductase activity is required for the clearance of membrane-tethered faulty gene products from the ER [18].
In addition to its role as topology-selective reductase in the folding and degradation pathways [17][18], palmitoylated TMX1 localizes at ER-mitochondria contact sites, aka MAM (mitochondria-associated membranes), to regulate Ca2+ flux between ER and mitochondria [31].
3. TMX2 and its Cytosolic Active Site
Among TMX family members, TMX2 (alternative name TXNDC14) undoubtedly is the most mysterious. It is a non-glycosylated protein of 296 amino acids, which has been identified in 2003 upon cloning from a fetal cDNA library [32]. The topology of TMX2 has been only recently characterized. Initially, it has been described as a type I membrane protein [32]. A recent study clarified its topology showing that TMX2 is a multi-spanning protein displaying both the N- and the C-terminal regions within the cytosolic side [12] (Figure 1). As such, the peculiar SNDC catalytic site of TMX2 is oriented towards the cytosol [12], in contrast with the other members of the TMX family (Figure 1). The long C-terminal tail of TMX2 contains a canonical ER retention signal (-KKDK) [32] (Figure 1). TMX2 is localized in different ER sub-compartments, such as in the nuclear outer membrane [12], or at the MAM [22]. TMX2 expression is ubiquitous with the highest levels in brain, heart, liver, kidney and pancreas [32]. Moreover, TMX2 is up-regulated upon oxidative stress, but not upon hypoxia, heat shock, or ER stress, consistently with the lack of an ERSE motif within its promoter region [12]. TMX2 gene deletion in mice is embryonic lethal, implying a crucial role of this protein at early stages of development [33]. So far, no information is available on the physiologic function of TMX2 (Table 1) with the exception of a possible participation of TMX2 in the importin-β:Ran complex that controls nuclear targeting of select cargo proteins [12][34]. The localization of TMX2 in the outer nuclear membrane, which is contiguous to the ER membrane, is crucial for the binding of the importin-β:Ran complex and the maintenance of the nucleocytoplasmic Ran protein gradient, whereas mutations in the TMX2 active site only partially impair them [12]. TMX2 can also localize at the MAM [22], where it forms a functional complex with CNX and the Ca2+-pump SERCA2 to modulate the calcium flux [33] in a function that mimics the one reported for TMX1 [35] and that could be exerted in tissue-specific manner. Consistently with its high expression in brain tissue, missense mutations of TMX2 have recently been associated with brain developmental abnormalities [33] and microlissencephaly [36], a rare congenital brain disorder [37]. This phenotype could result from a loss of the protective role of TMX2 from ER stresses [38], which represents an important concurrent cause of neuronal death [39].
4. TMX3, a Classic PDI
TMX3 is a single-pass type I glycoprotein of 454 amino acids (Figure 1), which has been identified in 2005 among the uncharacterized proteins containing the consensus sequence for a TRX-like domain [13]. This PDI family member, also known as TXNDC10, displays within its large N-terminal region two N-glycosylation sites (Figure 1) and three TRX-like domains, one catalytically active type-a domain followed by two inactive type-b (b and b′) domains [40]. Additionally, its C-terminal region contains a classical KKKD retention sequence [13] (Figure 1). TMX3 transcripts have been found in a great variety of tissues with the highest levels in heart and skeletal muscle [13]. Consistent with other TMX family members, TMX3 does not contain an ERSE motif and it is not upregulated upon ER stress [13]. The type-a TRX-like domain of TMX3 is characterized by a canonical CGHC sequence [13] (Figure 1 and Table 1), which corresponds to the catalytic sequence of PDI [41]. TMX3 acts as an oxidase in vitro [13] and its b′ domain is possibly involved in substrate recruitment [40]. The precise role of TMX3 in cells has not been established, yet. Preliminary studies show a protective function of TMX3 against neuronal atrophy in mice models for Huntington's disease [42], a progressive brain disorder caused by an inherited CAG trinucleotide repeat expansion in the huntingtin (HTT) gene [43]. The molecular basis of this protective effect is unclear, also because HTT is a cytosolic protein and a direct interaction with the functional portion of TMX3 can be ruled out. Since it has been shown that the expression of mutated HTT triggers ER stress, an hypothesis is that TMX3 protects cells against neuronal atrophy mitigating the stress situation [42]. Both deletion and missense mutations in TMX3 gene have been linked to coronary artery diseases [44] and microphthalmia [45], a disease associated with retarded growth of the eye [46].
5. TMX4, the Paralogue of TMX1
TMX4 (alternative name TXNDC13) is a single-pass type I glycoprotein (Figure 1) of 349 amino acids that has been identified in 2010 during a database search for TRX-like domain containing proteins [14]. Phylogenetic analysis showed that TMX4 represents the paralogue of TMX1 [21], with whom it shares high similarity within the N-terminal luminal regions despite the presence of an N-glycosylation site [14][21] and a di-arginine RQR retention motif within the C-terminal domain [21]. Both proteins display two phosphorylation sites within the cytosolic domain [23] (Figure 1), which could modulate sub-ER localization upon recruitment of select binding partners [21]. TMX4 expression is ubiquitous with the highest levels in heart tissue [14]. Consistently with the lack of an ERSE motif within its promoter region, TMX4 is not up-regulated during ER stresses [14]. TMX4 has one luminal type-a TRX-like domain, which contains a non-canonical CPSC active site [14] (Figure 1 and Table 1). The proline in position 2 hints at a role as ER-reductase [27] and, indeed, TMX4 efficiently reduces insulin disulfides in vitro [14]. Additionally, also TMX4 works as VKOR redox partner, even if this interaction results weaker compared to the one established by VKOR and TMX1 [30]. From the functional point of view, no evidence has been reported on substrate preference and possible roles of TMX4 in cells, even though different hypothesis have been formulated. The structural similarities with TMX1 strongly hint at a role of TMX4 as ER reductase possibly acting in both protein folding and degradation pathways.Finally, a recent study showed that TMX4 also distributes at the inner membrane of the nuclear envelope (NE) with the N-terminal portion facing the NE lumen [47]. Since many protein complexes resident at the NE (including the LINC:Torsin A complex) are modulated by redox cycles, a role for TMX4 in the regulation of the NE structure can be envisioned [47].
6. TMX5, a Natural Trapping Mutant Member of the TMX Family
TMX5 (TXNDC15) is another poorly characterized member of the TMX family. It is a predicted single-span type I protein of 360 amino acids (Figure 1). It has been identified in a large scale protein screening in 2003 [48]. Structurally, TMX5 has a large N-terminal luminal domain and a very short C-terminal cytoplasmic tail of 18 amino acids, which lacks canonical ER retention signals. This characteristic suggests that unlike other TMXs, TMX5 could potentially leave the ER and traffic through the secretory pathway. Unfortunately, no information is currently available on tissue expression and transcriptional regulation of TMX5. TMX5 possesses four putative N-glycosylation sites and one type-a TRX-like domain within its N-terminal luminal portion. The core of its TRX-like domain is represented by a non-canonical CRFS active site. The absence of the C-terminal cysteine residue within its active site defines TMX5 as a natural trapping mutant protein [49]. Indeed, the N-terminal cysteine of the TMX5 active site can nucleophilically attack free thiol group in protein substrates, but the interaction can be resolved only by the intervention of an external cysteine provided by another PDI or by the protein substrate itself. As such, the mixed disulfide between TMX5 and client proteins results stabilized and it could be long-living.
To date, no evidence has been reported on the physiologic roles of TMX5. Mutations in TMX5 gene have recently been associated with the development of the Meckel-Gruber syndrome (MKS), a rare perinatally lethal autosomal recessive disease caused by defective ciliogenesis [50]. Deletions and missense mutations result in the generation of truncated forms of TMX5 that do not localize within primary cilium or periciliary regions as the wild type [51][52][53]. Thus, the mis-localization or the premature degradation of TMX5 might correlate with the onset of such ciliopathies. Indeed, it has been found that patients' derived mutated fibroblasts as well as cells subjected to siRNA knockdown have a reduced number of ciliated cells, abnormal ciliary morphology and an aberrant localization to the transition zone of TMEM67 [51][52], a crucial regulator of cilia function [54].
7. Conclusion
We recapitulate the current knowledge about the features and roles of the members of the TMX family. These are five membrane-tethered PDIs, which are characterized by an ER signal sequence and a type-a TRX-like domain. Despite their similarities, the TMXs also show some structural differences, which could hint at a certain degree of functional diversification and specialization among the different members of the TMX family. Future dissection of their individual roles are needed to enlarge our knowledge about PDIs functions, and to allow the comparison between the members of the same TMX family and between membrane-tethered and soluble PDIs.
References
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419.
- Ellgaard, L.; McCaul, N.; Chatsisvili, A.; Braakman, I. Co- and Post-Translational Protein Folding in the ER. Traffic 2016, 17, 615–638.
- Kosuri, P.; Alegre-Cebollada, J.; Feng, J.; Kaplan, A.; Ingles-Prieto, A.; Badilla, C.L.; Stockwell, B.R.; Sanchez-Ruiz, J.M.; Holmgren, A.; Fernandez, J.M. Protein folding drives disulfide formation. Cell 2012, 151, 794–806.
- Hatahet, F.; Ruddock, L.W. Protein disulfide isomerase: A critical evaluation of its function in disulfide bond formation. Antioxid. Redox Signal. 2009, 11, 2807–2850.
- Pisoni, G.B.; Molinari, M. Five Questions (with their Answers) on ER-Associated Degradation. Traffic 2016, 17, 341–350.
- Suzuki, Y.; Schmitt, M.J. Redox diversity in ERAD-mediated protein retrotranslocation from the endoplasmic reticulum: A complex puzzle. Biol. Chem. 2015, 396, 539–554.
- Appenzeller-Herzog, C.; Ellgaard, L. The human PDI family: Versatility packed into a single fold. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2008, 1783, 535–548.
- Myllyharju, J. Prolyl 4-hydroxylases, the key enzymes of collagen biosynthesis. Matrix Biol. 2003, 22, 15–24.
- Shrimal, S.; Gilmore, R. Oligosaccharyltransferase structures provide novel insight into the mechanism of asparagine-linked glycosylation in prokaryotic and eukaryotic cells. Glycobiology 2019, 29, 288–297.
- Okumura, M.; Kadokura, H.; Inaba, K. Structures and functions of protein disulfide isomerase family members involved in proteostasis in the endoplasmic reticulum. Free Radic. Biol. Med. 2015, 83, 314–322.
- Hatahet, F.; Ruddock, L.W. Substrate recognition by the protein disulfide isomerases. FEBS J. 2007, 274, 5223–5234.
- Oguro, A.; Imaoka, S. Thioredoxin-related transmembrane protein 2 (TMX2) regulates the Ran protein gradient and importin-beta-dependent nuclear cargo transport. Sci. Rep. 2019, 9, 15296.
- Haugstetter, J.; Blicher, T.; Ellgaard, L. Identification and characterization of a novel thioredoxin-related transmembrane protein of the endoplasmic reticulum. J. Biol. Chem. 2005, 280, 8371–8380.
- Sugiura, Y.; Araki, K.; Iemura, S.; Natsume, T.; Hoseki, J.; Nagata, K. Novel thioredoxin-related transmembrane protein TMX4 has reductase activity. J. Biol. Chem. 2010, 285, 7135–7142.
- Kozlov, G.; Maattanen, P.; Thomas, D.Y.; Gehring, K. A structural overview of the PDI family of proteins. FEBS J. 2010, 277, 3924–3936.
- Matsuo, Y.; Akiyama, N.; Nakamura, H.; Yodoi, J.; Noda, M.; Kizaka-Kondoh, S. Identification of a novel thioredoxin-related transmembrane protein. J. Biol. Chem. 2001, 276, 10032–10038.
- Pisoni, G.B.; Ruddock, L.W.; Bulleid, N.; Molinari, M. Division of labor among oxidoreductases: TMX1 preferentially acts on transmembrane polypeptides. Mol. Biol. Cell 2015, 26, 3390–3400.
- Guerra, C.; Brambilla Pisoni, G.; Solda, T.; Molinari, M. The reductase TMX1 contributes to ERAD by preferentially acting on membrane-associated folding-defective polypeptides. Biochem. Biophys. Res. Commun. 2018, 503, 938–943.
- Akiyama, N.; Matsuo, Y.; Sai, H.; Noda, M.; Kizaka-Kondoh, S. Identification of a series of transforming growth factor beta-responsive genes by retrovirus-mediated gene trap screening. Mol. Cell Biol. 2000, 20, 3266–3273.
- Matsuo, Y.; Nishinaka, Y.; Suzuki, S.; Kojima, M.; Kizaka-Kondoh, S.; Kondo, N.; Son, A.; Sakakura-Nishiyama, J.; Yamaguchi, Y.; Masutani, H.; et al. A human transmembrane oxidoreductase of the thioredoxin family: The possible role in disulfide-linked protein folding in the endoplasmic reticulum. Arch. Biochem. Biophys. 2004, 423, 81–87.
- Roth, D.; Lynes, E.; Riemer, J.; Hansen, H.G.; Althaus, N.; Simmen, T.; Ellgaard, L. A di-arginine motif contributes to the ER localization of the type I transmembrane ER oxidoreductase TMX4. Biochem. J. 2009, 425, 195–205.
- Lynes, E.M.; Bui, M.; Yap, M.C.; Benson, M.D.; Schneider, B.; Ellgaard, L.; Berthiaume, L.G.; Simmen, T. Palmitoylated TMX and calnexin target to the mitochondria-associated membrane. EMBO J. 2012, 31, 457–470.
- Olsen, J.V.; Blagoev, B.; Gnad, F.; Macek, B.; Kumar, C.; Mortensen, P.; Mann, M. Global, In Vivo, and Site-Specific Phosphorylation Dynamics in Signaling Networks. Cell 2006, 127, 635–648.
- Galligan, J.J.; Petersen, D.R. The human protein disulfide isomerase gene family. Hum. Genom. 2012, 6, 6.
- Matsuo, Y.; Masutani, H.; Son, A.; Kizaka-Kondoh, S.; Yodoi, J. Physical and functional interaction of transmembrane thioredoxin-related protein with major histocompatibility complex class I heavy chain: Redox-based protein quality control and its potential relevance to immune responses. Mol. Biol. Cell 2009, 20, 4552–4562.
- Matsuo, Y.; Irie, K.; Kiyonari, H.; Okuyama, H.; Nakamura, H.; Son, A.; Lopez-Ramos, D.A.; Tian, H.; Oka, S.; Okawa, K.; et al. The protective role of the transmembrane thioredoxin-related protein TMX in inflammatory liver injury. Antioxid. Redox Signal. 2013, 18, 1263–1272.
- Hatahet, F.; Ruddock, L.W. Modulating proteostasis: Peptidomimetic inhibitors and activators of protein folding. Curr. Pharm. Des. 2009, 15, 2488–2507.
- Roos, G.; Garcia-Pino, A.; Van belle, K.; Brosens, E.; Wahni, K.; Vandenbussche, G.; Wyns, L.; Loris, R.; Messens, J. The Conserved Active Site Proline Determines the Reducing Power of Staphylococcus aureus Thioredoxin. J. Mol. Biol. 2007, 368, 800–811.
- Pasetto, M.; Barison, E.; Castagna, M.; Della Cristina, P.; Anselmi, C.; Colombatti, M. Reductive activation of type 2 ribosome-inactivating proteins is promoted by transmembrane thioredoxin-related protein. J. Biol. Chem. 2012, 287, 7367–7373.
- Schulman, S.; Wang, B.; Li, W.; Rapoport, T.A. Vitamin K epoxide reductase prefers ER membrane-anchored thioredoxin-like redox partners. Proc. Natl. Acad. Sci. USA 2010, 107, 15027–15032.
- Janssens, S.; Bultynck, G.; Krols, M. ER–Mitochondria contact sites: A new regulator of cellular calcium flux comes into play. J. Cell Biol. 2016, 214, 367–370.
- Meng, X.; Zhang, C.; Chen, J.; Peng, S.; Cao, Y.; Ying, K.; Xie, Y.; Mao, Y. Cloning and Identification of a Novel cDNA Coding Thioredoxin-Related Transmembrane Protein 2. Biochem. Genet. 2003, 41, 99–106.
- Vandervore, L.V.; Schot, R.; Milanese, C.; Smits, D.J.; Kasteleijn, E.; Fry, A.E.; Pilz, D.T.; Brock, S.; Borklu-Yucel, E.; Post, M.; et al. TMX2 Is a Crucial Regulator of Cellular Redox State, and Its Dysfunction Causes Severe Brain Developmental Abnormalities. Am. J. Hum. Genet. 2019, 105, 1126–1147.
- Quimby, B.B.; Corbett, A.H. Nuclear transport mechanisms. Cell. Mol. Life Sci. 2001, 58, 1766–1773.
- Raturi, A.; Gutierrez, T.; Ortiz-Sandoval, C.; Ruangkittisakul, A.; Herrera-Cruz, M.S.; Rockley, J.P.; Gesson, K.; Ourdev, D.; Lou, P.H.; Lucchinetti, E.; et al. TMX1 determines cancer cell metabolism as a thiol-based modulator of ER-mitochondria Ca2+ flux. J. Cell Biol. 2016, 214, 433–444.
- Ghosh, S.G.; Wang, L.; Breuss, M.W.; Green, J.D.; Stanley, V.; Yang, X.; Ross, D.; Traynor, B.J.; Alhashem, A.M.; Azam, M.; et al. Recurrent homozygous damaging mutation in TMX2, encoding a protein disulfide isomerase, in four families with microlissencephaly. J. Med. Genet. 2020, 57, 274–282.
- Fry, A.E.; Cushion, T.D.; Pilz, D.T. The genetics of lissencephaly. Am. J. Med. Genet. C Semin. Med. Genet. 2014, 166, 198–210.
- Kramer, N.J.; Haney, M.S.; Morgens, D.W.; Jovičić, A.; Couthouis, J.; Li, A.; Ousey, J.; Ma, R.; Bieri, G.; Tsui, C.K.; et al. CRISPR–Cas9 screens in human cells and primary neurons identify modifiers of C9ORF72 dipeptide-repeat-protein toxicity. Nat. Genet. 2018, 50, 603–612.
- Poulton, C.J.; Schot, R.; Kia, S.K.; Jones, M.; Verheijen, F.W.; Venselaar, H.; de Wit, M.-C.Y.; de Graaff, E.; Bertoli-Avella, A.M.; Mancini, G.M.S. Microcephaly with Simplified Gyration, Epilepsy, and Infantile Diabetes Linked to Inappropriate Apoptosis of Neural Progenitors. Am. J. Hum. Genet. 2011, 89, 265–276.
- Haugstetter, J.; Maurer, M.A.; Blicher, T.; Pagac, M.; Wider, G.; Ellgaard, L. Structure-Function Analysis of the Endoplasmic Reticulum Oxidoreductase TMX3 Reveals Interdomain Stabilization of the N-terminal Redox-active Domain. J. Biol. Chem. 2007, 282, 33859–33867.
- Wilkinson, B.; Gilbert, H.F. Protein disulfide isomerase. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2004, 1699, 35–44.
- Fox, J.; Lu, Z.; Barrows, L. Thiol-disulfide Oxidoreductases TRX1 and TMX3 Decrease Neuronal Atrophy in a Lentiviral Mouse Model of Huntington’s Disease. PLoS Curr. 2015.
- McColgan, P.; Tabrizi, S.J. Huntington’s disease: A clinical review. Eur. J. Neurol. 2018, 25, 24–34.
- Li, M.; Wen, Y.; Wen, H.; Gui, C.; Huang, F.; Zeng, Z. Discovery of PPP2R3A and TMX3 pathogenic variants in a Zhuang family with coronary artery disease using whole-exome sequencing. Int. J. Clin. Exp. Pathol. 2018, 11, 3678–3684.
- Abraham, E.; Chao, R.; Nevin, L.; Agarwal, P.; Riemer, J.; Bai, X.; Delaney, A.; Akana, M.; JimenezLopez, N.; Bardakjian, T.; et al. A Male with Unilateral Microphthalmia Reveals a Role for TMX3 in Eye Development. PLoS ONE 2010, 5, e10565.
- Verma, A.S.; Fitzpatrick, D.R. Anophthalmia and microphthalmia. Orphanet J. Rare Dis. 2007, 2, 47.
- Cheng, L.C.; Baboo, S.; Lindsay, C.; Brusman, L.; Martinez-Bartolome, S.; Tapia, O.; Zhang, X.; Yates, J.R., 3rd; Gerace, L. Identification of new transmembrane proteins concentrated at the nuclear envelope using organellar proteomics of mesenchymal cells. Nucleus 2019, 10, 126–143.
- Clark, H.F. The Secreted Protein Discovery Initiative (SPDI), a Large-Scale Effort to Identify Novel Human Secreted and Transmembrane Proteins: A Bioinformatics Assessment. Genome Res. 2003, 13, 2265–2270.
- Yang, K.; Li, D.F.; Wang, X.; Liang, J.; Sitia, R.; Wang, C.C.; Wang, X. Crystal Structure of the ERp44-Peroxiredoxin 4 Complex Reveals the Molecular Mechanisms of Thiol-Mediated Protein Retention. Structure 2016, 24, 1755–1765.
- Hartill, V.; Szymanska, K.; Sharif, S.M.; Wheway, G.; Johnson, C.A. Meckel–Gruber Syndrome: An Update on Diagnosis, Clinical Management, and Research Advances. Front. Pediatrics 2017, 5, 244.
- Shaheen, R.; Szymanska, K.; Basu, B.; Patel, N.; Ewida, N.; Faqeih, E.; Al Hashem, A.; Derar, N.; Alsharif, H.; Aldahmesh, M.A.; et al. Characterizing the morbid genome of ciliopathies. Genome Biol. 2016, 17, 1–11.
- Radhakrishnan, P.; Nayak, S.S.; Shukla, A.; Lindstrand, A.; Girisha, K.M. Meckel syndrome: Clinical and mutation profile in six fetuses. Clin. Genet. 2019, 96, 560–565.
- Ridnõi, K.; Šois, M.; Vaidla, E.; Pajusalu, S.; Kelder, L.; Reimand, T.; Õunap, K. A prenatally diagnosed case of Meckel–Gruber syndrome with novel compound heterozygous pathogenic variants in the.TXNDC15. gene. Mol. Genet. Genom. Med. 2019, 7, e614.
- Leightner, A.C.; Hommerding, C.J.; Peng, Y.; Salisbury, J.L.; Gainullin, V.G.; Czarnecki, P.G.; Sussman, C.R.; Harris, P.C. The Meckel syndrome protein meckelin (TMEM67) is a key regulator of cilia function but is not required for tissue planar polarity. Hum. Mol. Genet. 2013, 22, 2024–2040.