Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Conner Chen and Version 1 by Canhua Huang.
Reactive oxygen species (ROS) are a class of highly reactive free radicals, such as hydroxyl radical (•OH), the superoxide radical (O2•−) and hydrogen peroxide (H2O2). The high intracellular ROS level-induced oxidative stress leads to the upregulation of antioxidant capacity to maintain redox homeostasis by metabolic rerouting or activation of genetic programs.
- reactive oxygen species
- oxidative stress
- cancer therapy
1. Introduction
Redox homeostasis is essential for biological function and its disturbance leads to profound pathophysiological consequences in cells, which emphasize the balance between the relative abundance of reactive oxygen species (ROS) and antioxidants [1][2][3]. However, cells may generate excessive ROS as an unavoidable result of alterations in metabolic signaling pathways [4][5]. Oxidative stress arises when ROS are excessively produced, while antioxidants are relatively insufficient. The ROS levels are tightly regulated by antioxidant systems, including enzymatic antioxidant and nonenzymatic antioxidant systems. To accommodate oxidative stress, cells modify metabolic and genetic reprogramming, thereby leading to increased production of NADPH, glutathione (GSH, l-γ-glutamyl-l-cysteinyl-glycine), superoxide dismutases (SODs) and thioredoxins (TRXs), returning ROS to homeostatic levels [6][7][8].
When the high ROS level exceeds non-toxic doses, ROS may cause oxidative damage to macromolecules, such as nucleic acids, proteins, lipids and glucose, resulting in fragmentation of enzymes and structural proteins, membrane damage, gene mutations and even pro-oncogenic signaling activation [9][10]. Increased oxidative stress can initiate tumor development and contribute to tumor progression by directly oxidizing macromolecules or oxidative stress-caused aberrant redox signaling [11], demonstrating that high ROS levels may increase the risk of cancer when antioxidant systems are insufficient to protect cells from oxidative stress. Since oxidative stress plays an important role in carcinogenesis and cancer progression [2][12][13], it is an attractive idea to use antioxidants for the treatment of cancer. Numerous antioxidants were developed in the past few decades. They can be classified as nonenzymatic antioxidants, such as NF-E2 p45-related factor 2 (NRF2) activators [14], vitamins [15], N-acetylcysteine (NAC) and GSH esters [16][17], and enzymatic antioxidants, such as NADPH oxidase (NOX) inhibitors [18] and SOD mimics [19]. Some of them have shown potential to act as anticancer drugs and multiple antioxidant therapeutic strategies were explored in pre-clinical and clinical research [20].
2. Redox Homeostasis: The Biological Basis for Antioxidant Therapy
ROS are a class of highly reactive free radicals, such as hydroxyl radical (•OH), the superoxide radical (O2•−) and hydrogen peroxide (H2O2) [21][22]. The high intracellular ROS level-induced oxidative stress leads to the upregulation of antioxidant capacity to maintain redox homeostasis by metabolic rerouting or activation of genetic programs [23][24]. Disruption of redox homeostasis contributes to multiple human diseases, including cancer, and resetting redox homeostasis with antioxidants is a promising strategy to prevent tumorigenesis or inhibit cancer progression. It is well known that redox homeostasis is balanced by the equilibrium of ROS generation and ROS elimination. Therefore, we first describe the underlying mechanisms that regulate the cellular redox homeostasis would be first described (Figure 1).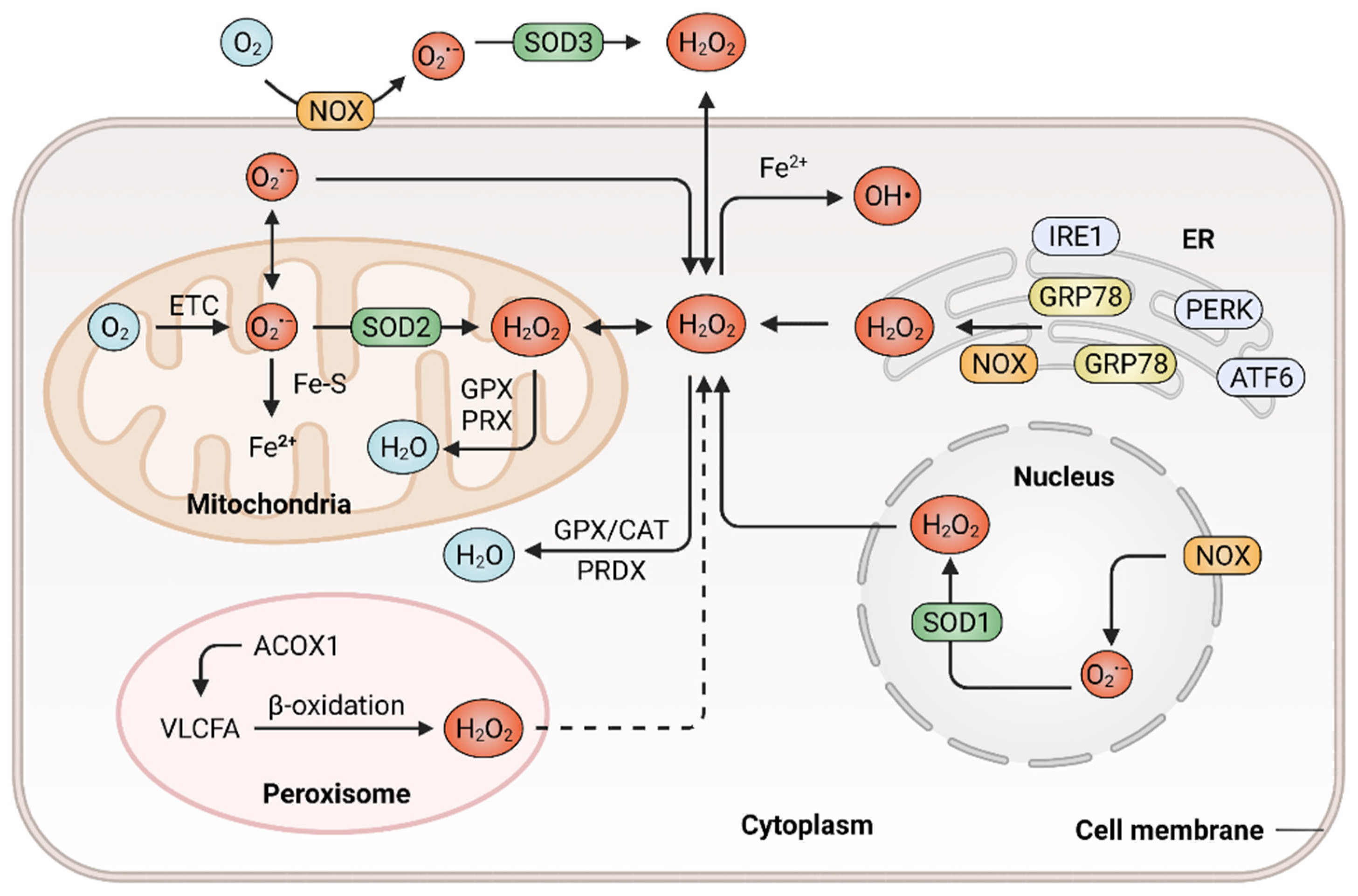
Figure 1. Generation and elimination of ROS in mammalian cells. ROS are generated extracellularly by NADPH oxidase (NOX) or intracellularly in different subcellular compartments, including endoplasmic reticulum (ER), peroxisome, nucleus as well as the mitochondrial electron transport chain (ETC). Antioxidant systems include the peroxiredoxin (PRDX), the glutathione peroxidase (GPX) and catalase (CAT) in the cytosol or mitochondria, which hydrolyze H2O2 to H2O.
2.1. Mechanisms in ROS Generation
ROS are prominently generated by transmembrane NOXs and other various oxidases from the mitochondrial electron transport chain (ETC) [25], endoplasmic reticulum (ER) [26] and peroxisomes [27], in response to intracellular signaling and extracellular stimuli. The mitochondrion functions as a highly dynamic organelle and an essential endogenous enzymatic source of ROS, which generates ROS through ETC, a series of electron transfer complexes located on the mitochondrial inner membrane [28][29]. The production of mitochondrial ROS is associated with the metabolism of glucose, fatty acids and amino acids (via glycolysis, β-oxidation and oxidative deamination, respectively), which provide precursors for tricarboxylic acid (TCA) cycle to produce metabolic substrates that enter the ETC [30][31]. In the mitochondrial ETC, ROS generation is probably due to the leak of electrons from complex I, II and III. During this process, oxygen is reduced with a single electron and thus generating O2•−, which can be dismutated to H2O2 [32][33]. The rate of ROS generation from the mitochondrial ETC is predominantly dependent on the concentration of the one-electron donor and the reaction rate between the donor and oxygen. The primary function of NOXs is to produce ROS, which is triggered by a variety of factors and reported to be associated with tumor development [34]. The NOX family consists of seven members, namely NOX1, NOX2, NOX3, NOX4, NOX5, DUOX1 and DUOX2 [35]. They catalyze the conversion of oxygen to O2•− by transferring electrons to molecular oxygen in various subcellular compartments, such as the nucleus [36]. NOXs-derived ROS might activate the downstream secondary oxidase systems, such as xanthine oxidase and uncoupled endothelial nitric oxide synthase, further aggravating oxidative stress and accelerating the development of cancer [37]. ER is a protein-folding factory, which plays an important role in normal physiology [38][39]. The oxidizing site in ER supports the proper conformation and post-translational modifications of nascent proteins [40]. In response to the aggregation of unfolded or misfolded proteins within ER, glucose-regulated protein 78 (GRP78) dissociates from ER stress receptors, such as activating transcription factor 6 (ATF6), pancreatic ER kinase (PKR)-like ER kinase (PERK) and inositol-requiring enzyme 1 (IRE1), leading to ER stress and eventually resulting in ROS accumulation [41][42][43]. In addition, the release of calcium from the ER and depolarization of the mitochondrial inner membrane can stimulate the production of mitochondrial ROS and mediate excessive oxidative stress [44][45]. As multifunctional dynamic organelles, peroxisomes exist in almost all eukaryotic cells and play essential roles in redox homeostasis [46][47]. The name of peroxisomes derives from their function in the metabolism of H2O2 [27]. Peroxisomal respiration accounts for approximately 20% of total oxygen consumption and produces up to 35% of total H2O2 by peroxisomal oxidases in certain mammalian cells [48]. For instance, peroxisomal oxidase acyl-CoA oxidase 1 (ACOX1), the rate-limiting enzyme in fatty acid β-oxidation, can oxidize very long-chain fatty acid (VLCFA) and lead to H2O2 production in peroxisomes. In addition, a gain-of-function mutation in ACOX1 may further enhance the ROS levels [49]. Besides, the ACOX1-induction of ROS production was demonstrated to be involved in oxidative DNA damage and the progression of hepatocellular carcinoma (HCC) [50]. Ultraviolet (UV) radiation is also an important factor that contributes to ROS generation and subsequent carcinogenesis [51]. Cells exhibit an increased production of ROS when exposed to UV radiation. UV-induced transition-type mutations at dipyrimidine sites frequently occur in the RAS oncogene and p53 tumor suppressor gene [52]. In addition, a wide range of biological phenomena, such as inflammatory and oxidative modifications of macromolecules, were reported to participate in UV-induced skin carcinogenesis and the progression of glioblastoma [53][54].2.2. ROS Elimination with Enzymatic or Nonenzymatic Antioxidant System
Increased accumulation of ROS can be eliminated by various enzymatic antioxidant systems including SODs [55], GSH peroxidases (GPXs) [56], peroxiredoxins (PRDXs) [57], paraoxonase (PONs) and catalase (CAT) [58]. Additionally, ROS can also be eliminated by nonenzymatic antioxidant systems, such as GSH [59] and TRXs [60]. The antioxidant systems counteract ROS-mediated damage to maintain ROS homeostasis, enabling tumor cell survival [20]. The enzymatic antioxidant system mainly consists of SODs, PRDXs, CAT, PONs and GPXs. Under oxidative stress, these antioxidant enzymes are upregulated or activated to prevent oxidative damage. SODs catalyze the conversion of O2•− into molecular oxygen and H2O2, thus controlling the levels of ROS and limiting their potential toxicity [61]. Since SOD1 was firstly discovered in 1969, all of the three members in the SOD family were biochemically and molecularly characterized in mammalian cells, including Cu/Zn-SOD (SOD1), Mn-SOD (SOD2) and EC-SOD (SOD3) [62]. SOD1 and SOD2 localize in cytosol, the mitochondrial inter membrane space, the nucleus and the mitochondrial matrix, while SOD3 is secreted into the extracellular space [63][64][65]. The SOD family constitutes the first line of defense against ROS. The O2•− is dismutated by SODs to form H2O2, which can be decomposed into O2 and H2O by CAT or GPXs [66]. Several enzymes, such as glutathione reductase and glucose-6-phosphate dehydrogenase, function as secondary antioxidant enzymes that enable GPX to function with cofactors (NADPH, GSH and glucose 6-phosphate) but not to act on ROS directly [67][68]. PON2 is one member of the PON family that consists of three members, namely PON1, PON2 and PON3. PON2 is a membrane-associated protein that is located in the plasma membrane, mitochondria and ER [69]. It was reported that PON2 protects against oxidative stress, both in vivo and in vitro [70][71]. For instance, PON2 binds with high affinity to coenzyme Q10 and protects against mitochondrial dysfunction when localized to the mitochondria, while PON2 deficiency results in mitochondrial oxidative stress [72]. Nonenzymatic antioxidants are non-catalytic small molecules that can quench ROS and reduce oxidative stress [73]. The most abundant nonenzymatic antioxidant is GSH, a tripeptide composed of glutamate, cysteine and glycine. Its synthesis is regulated by glutaminases (GLS1 and GLS2), the cystine-glutamate antiporter xCT (SLC7A11), the GSH biosynthetic rate-limiting enzyme glutamate-cysteine ligase (GCL) and the GSH synthetase (GSS) [74][75]. GCL is a heterodimeric holoenzyme that is composed of catalytic (GCLC) and modifier (GCLM) subunits; the expression levels of GCLC and GCLM are highly associated with the drug sensitivity of cancer cells and patient survival [76]. Moreover, the silencing of SLC7A11, GCLC and GSS represses the proliferation of clear cell renal cell carcinoma by decreasing the cellular GSH levels. However, reduced levels of GSH were also observed in patients with breast or colon cancers, especially in the advanced stages of these diseases, indicating the essential role of GSH in cancer cell survival [77][78]. Another nonenzymatic antioxidant is the TRX system, which is composed of TRXs and NADPH-dependent thioredoxin reductase (TrxR), which participate in the removal of harmful and excessive H2O2 [79]. There are two kinds of TRXs in mammalian cells, known as cytosolic TRX1 and mitochondrial TRX2 [80]. TRXs directly donate electrons to thiol-dependent PRDXs to remove H2O2. Oxidized TRXs are then reduced by TrxR, with NADPH as a cofactor [81]. Moreover, the oxidized PRDXs can also be reduced by TRXs [57]. Given the important role of the TRX system in cellular redox homeostasis, disturbance in the TRXs’ metabolism is highly associated with the progression and chemoresistance of multiple tumors [82], thus making TRXs essential targets for anticancer therapy.References
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709.
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197.
- Peng, L.; Jiang, J.; Chen, H.N.; Zhou, L.; Huang, Z.; Qin, S.; Jin, P.; Luo, M.; Li, B.; Shi, J.; et al. Redox-sensitive cyclophilin A elicits chemoresistance through realigning cellular oxidative status in colorectal cancer. Cell Rep. 2021, 37, 110069.
- Wang, K.; Jiang, J.; Lei, Y.; Zhou, S.; Wei, Y.; Huang, C. Targeting Metabolic-Redox Circuits for Cancer Therapy. Trends Biochem. Sci. 2019, 44, 401–414.
- Liang, D.; Minikes, A.M.; Jiang, X. Ferroptosis at the intersection of lipid metabolism and cellular signaling. Mol. Cell 2022.
- Virag, L.; Jaen, R.I.; Regdon, Z.; Bosca, L.; Prieto, P. Self-defense of macrophages against oxidative injury: Fighting for their own survival. Redox Biol. 2019, 26, 101261.
- Caserta, S.; Ghezzi, P. Release of redox enzymes and micro-RNAs in extracellular vesicles, during infection and inflammation. Free Radic. Biol. Med. 2021, 169, 248–257.
- Gonzalez-Chavez, Z.; Vazquez, C.; Mejia-Tlachi, M.; Marquez-Duenas, C.; Manning-Cela, R.; Encalada, R.; Rodriguez-Enriquez, S.; Michels, P.A.M.; Moreno-Sanchez, R.; Saavedra, E. Gamma-glutamylcysteine synthetase and tryparedoxin 1 exert high control on the antioxidant system in Trypanosoma cruzi contributing to drug resistance and infectivity. Redox Biol. 2019, 26, 101231.
- Chaiswing, L.; St Clair, W.H.; St Clair, D.K. Redox Paradox: A Novel Approach to Therapeutics-Resistant Cancer. Antioxid. Redox Signal. 2018, 29, 1237–1272.
- Urso, L.; Cavallari, I.; Sharova, E.; Ciccarese, F.; Pasello, G.; Ciminale, V. Metabolic rewiring and redox alterations in malignant pleural mesothelioma. Br. J. Cancer 2020, 122, 52–61.
- Canli, O.; Nicolas, A.M.; Gupta, J.; Finkelmeier, F.; Goncharova, O.; Pesic, M.; Neumann, T.; Horst, D.; Lower, M.; Sahin, U.; et al. Myeloid Cell-Derived Reactive Oxygen Species Induce Epithelial Mutagenesis. Cancer Cell 2017, 32, 869–883.e865.
- Luo, M.; Huang, Z.; Yang, X.; Chen, Y.; Jiang, J.; Zhang, L.; Zhou, L.; Qin, S.; Jin, P.; Fu, S.; et al. PHLDB2 Mediates Cetuximab Resistance via Interacting With EGFR in Latent Metastasis of Colorectal Cancer. Cell Mol. Gastroenterol. Hepatol. 2022, 13, 1223–1242.
- Klaunig, J.E. Oxidative Stress and Cancer. Curr. Pharm. Des. 2018, 24, 4771–4778.
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43.
- Ngo, B.; Van Riper, J.M.; Cantley, L.C.; Yun, J. Targeting cancer vulnerabilities with high-dose vitamin C. Nat. Rev. Cancer 2019, 19, 271–282.
- Li, P.; Wu, M.; Wang, J.; Sui, Y.; Liu, S.; Shi, D. NAC selectively inhibit cancer telomerase activity: A higher redox homeostasis threshold exists in cancer cells. Redox Biol. 2016, 8, 91–97.
- Saidu, N.E.B.; Kavian, N.; Leroy, K.; Jacob, C.; Nicco, C.; Batteux, F.; Alexandre, J. Dimethyl fumarate, a two-edged drug: Current status and future directions. Med. Res. Rev. 2019, 39, 1923–1952.
- Augsburger, F.; Filippova, A.; Rasti, D.; Seredenina, T.; Lam, M.; Maghzal, G.; Mahiout, Z.; Jansen-Durr, P.; Knaus, U.G.; Doroshow, J.; et al. Pharmacological characterization of the seven human NOX isoforms and their inhibitors. Redox Biol. 2019, 26, 101272.
- Batinic-Haberle, I.; Tome, M.E. Thiol regulation by Mn porphyrins, commonly known as SOD mimics. Redox Biol. 2019, 25, 101139.
- Harris, I.S.; DeNicola, G.M. The Complex Interplay between Antioxidants and ROS in Cancer. Trends Cell Biol. 2020, 30, 440–451.
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383.
- Sies, H.; Belousov, V.V.; Chandel, N.S.; Davies, M.J.; Jones, D.P.; Mann, G.E.; Murphy, M.P.; Yamamoto, M.; Winterbourn, C. Defining roles of specific reactive oxygen species (ROS) in cell biology and physiology. Nat. Rev. Mol. Cell Biol. 2022, 1–17.
- Galan-Cobo, A.; Sitthideatphaiboon, P.; Qu, X.; Poteete, A.; Pisegna, M.A.; Tong, P.; Chen, P.H.; Boroughs, L.K.; Rodriguez, M.L.M.; Zhang, W.; et al. LKB1 and KEAP1/NRF2 Pathways Cooperatively Promote Metabolic Reprogramming with Enhanced Glutamine Dependence in KRAS-Mutant Lung Adenocarcinoma. Cancer Res. 2019, 79, 3251–3267.
- Wang, J.; Lu, Q.; Cai, J.; Wang, Y.; Lai, X.; Qiu, Y.; Huang, Y.; Ke, Q.; Zhang, Y.; Guan, Y.; et al. Nestin regulates cellular redox homeostasis in lung cancer through the Keap1-Nrf2 feedback loop. Nat. Commun. 2019, 10, 5043.
- Schofield, J.H.; Schafer, Z.T. Mitochondrial Reactive Oxygen Species and Mitophagy: A Complex and Nuanced Relationship. Antioxid. Redox Signal. 2021, 34, 517–530.
- Cantoni, O.; Zito, E.; Fiorani, M.; Guidarelli, A. Arsenite impinges on endoplasmic reticulum-mitochondria crosstalk to elicit mitochondrial ROS formation and downstream toxicity. Semin. Cancer Biol. 2021, 76, 132–138.
- He, A.; Dean, J.M.; Lodhi, I.J. Peroxisomes as cellular adaptors to metabolic and environmental stress. Trends Cell Biol. 2021, 31, 656–670.
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol. 2020, 37, 101674.
- Raimondi, V.; Ciccarese, F.; Ciminale, V. Oncogenic pathways and the electron transport chain: A dangeROS liaison. Br. J. Cancer 2020, 122, 168–181.
- Dambrova, M.; Zuurbier, C.J.; Borutaite, V.; Liepinsh, E.; Makrecka-Kuka, M. Energy substrate metabolism and mitochondrial oxidative stress in cardiac ischemia/reperfusion injury. Free Radic. Biol. Med. 2021, 165, 24–37.
- Flockhart, M.; Nilsson, L.C.; Tais, S.; Ekblom, B.; Apro, W.; Larsen, F.J. Excessive exercise training causes mitochondrial functional impairment and decreases glucose tolerance in healthy volunteers. Cell Metab. 2021, 33, 957–970.e956.
- Stefanatos, R.; Sanz, A. The role of mitochondrial ROS in the aging brain. FEBS Lett. 2018, 592, 743–758.
- Marin, R.; Chiarello, D.I.; Abad, C.; Rojas, D.; Toledo, F.; Sobrevia, L. Oxidative stress and mitochondrial dysfunction in early-onset and late-onset preeclampsia. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165961.
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313.
- Zhang, Y.; Murugesan, P.; Huang, K.; Cai, H. NADPH oxidases and oxidase crosstalk in cardiovascular diseases: Novel therapeutic targets. Nat. Rev. Cardiol. 2020, 17, 170–194.
- Magnani, F.; Mattevi, A. Structure and mechanisms of ROS generation by NADPH oxidases. Curr. Opin. Struct. Biol. 2019, 59, 91–97.
- Dang, P.M.; Rolas, L.; El-Benna, J. The Dual Role of Reactive Oxygen Species-Generating Nicotinamide Adenine Dinucleotide Phosphate Oxidases in Gastrointestinal Inflammation and Therapeutic Perspectives. Antioxid. Redox Signal. 2020, 33, 354–373.
- Sitia, R.; Braakman, I. Quality control in the endoplasmic reticulum protein factory. Nature 2003, 426, 891–894.
- Wadgaonkar, P.; Chen, F. Connections between endoplasmic reticulum stress-associated unfolded protein response, mitochondria, and autophagy in arsenic-induced carcinogenesis. Semin. Cancer Biol. 2021, 76, 258–266.
- Bibli, S.I.; Fleming, I. Oxidative Post-Translational Modifications: A Focus on Cysteine S-Sulfhydration and the Regulation of Endothelial Fitness. Antioxid. Redox Signal. 2021, 35, 1494–1514.
- Samanta, S.; Yang, S.; Debnath, B.; Xue, D.; Kuang, Y.; Ramkumar, K.; Lee, A.S.; Ljungman, M.; Neamati, N. The Hydroxyquinoline Analogue YUM70 Inhibits GRP78 to Induce ER Stress-Mediated Apoptosis in Pancreatic Cancer. Cancer Res. 2021, 81, 1883–1895.
- Ren, J.; Bi, Y.; Sowers, J.R.; Hetz, C.; Zhang, Y. Endoplasmic reticulum stress and unfolded protein response in cardiovascular diseases. Nat. Rev. Cardiol. 2021, 18, 499–521.
- Bhattarai, K.R.; Chaudhary, M.; Kim, H.R.; Chae, H.J. Endoplasmic Reticulum (ER) Stress Response Failure in Diseases. Trends Cell Biol. 2020, 30, 672–675.
- Wong, C.O.; Karagas, N.E.; Jung, J.; Wang, Q.; Rousseau, M.A.; Chao, Y.; Insolera, R.; Soppina, P.; Collins, C.A.; Zhou, Y.; et al. Regulation of longevity by depolarization-induced activation of PLC-beta-IP3R signaling in neurons. Proc. Natl. Acad. Sci. USA 2021, 118, e2004253118.
- Booth, D.M.; Varnai, P.; Joseph, S.K.; Hajnoczky, G. Oxidative bursts of single mitochondria mediate retrograde signaling toward the ER. Mol. Cell 2021, 81, 3866–3876.e3862.
- Fransen, M.; Lismont, C. Redox Signaling from and to Peroxisomes: Progress, Challenges, and Prospects. Antioxid. Redox Signal. 2019, 30, 95–112.
- Sargsyan, Y.; Thoms, S. Staying in Healthy Contact: How Peroxisomes Interact with Other Cell Organelles. Trends Mol. Med. 2020, 26, 201–214.
- Ivashchenko, O.; Van Veldhoven, P.P.; Brees, C.; Ho, Y.S.; Terlecky, S.R.; Fransen, M. Intraperoxisomal redox balance in mammalian cells: Oxidative stress and interorganellar cross-talk. Mol. Biol. Cell 2011, 22, 1440–1451.
- Chung, H.L.; Wangler, M.F.; Marcogliese, P.C.; Jo, J.; Ravenscroft, T.A.; Zuo, Z.; Duraine, L.; Sadeghzadeh, S.; Li-Kroeger, D.; Schmidt, R.E.; et al. Loss- or Gain-of-Function Mutations in ACOX1 Cause Axonal Loss via Different Mechanisms. Neuron 2020, 106, 589–606.e586.
- Chen, X.F.; Tian, M.X.; Sun, R.Q.; Zhang, M.L.; Zhou, L.S.; Jin, L.; Chen, L.L.; Zhou, W.J.; Duan, K.L.; Chen, Y.J.; et al. SIRT5 inhibits peroxisomal ACOX1 to prevent oxidative damage and is downregulated in liver cancer. EMBO Rep. 2018, 19, e45124.
- Emanuelli, M.; Sartini, D.; Molinelli, E.; Campagna, R.; Pozzi, V.; Salvolini, E.; Simonetti, O.; Campanati, A.; Offidani, A. The Double-Edged Sword of Oxidative Stress in Skin Damage and Melanoma: From Physiopathology to Therapeutical Approaches. Antioxidants 2022, 11, 612.
- Nishigori, C.; Hattori, Y.; Toyokuni, S. Role of reactive oxygen species in skin carcinogenesis. Antioxid. Redox Signal. 2004, 6, 561–570.
- Allouche, J.; Rachmin, I.; Adhikari, K.; Pardo, L.M.; Lee, J.H.; McConnell, A.M.; Kato, S.; Fan, S.; Kawakami, A.; Suita, Y.; et al. NNT mediates redox-dependent pigmentation via a UVB- and MITF-independent mechanism. Cell 2021, 184, 4268–4283.e4220.
- Kesanakurti, D.; Maddirela, D.; Banasavadi-Siddegowda, Y.K.; Lai, T.H.; Qamri, Z.; Jacob, N.K.; Sampath, D.; Mohanam, S.; Kaur, B.; Puduvalli, V.K. A novel interaction of PAK4 with PPARgamma to regulate Nox1 and radiation-induced epithelial-to-mesenchymal transition in glioma. Oncogene 2017, 36, 5309–5320.
- Wang, Y.; Branicky, R.; Noe, A.; Hekimi, S. Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J. Cell Biol. 2018, 217, 1915–1928.
- Shui, S.; Zhao, Z.; Wang, H.; Conrad, M.; Liu, G. Non-enzymatic lipid peroxidation initiated by photodynamic therapy drives a distinct ferroptosis-like cell death pathway. Redox Biol. 2021, 45, 102056.
- Bolduc, J.; Koruza, K.; Luo, T.; Malo Pueyo, J.; Vo, T.N.; Ezerina, D.; Messens, J. Peroxiredoxins wear many hats: Factors that fashion their peroxide sensing personalities. Redox Biol. 2021, 42, 101959.
- Patgiri, A.; Skinner, O.S.; Miyazaki, Y.; Schleifer, G.; Marutani, E.; Shah, H.; Sharma, R.; Goodman, R.P.; To, T.L.; Robert Bao, X.; et al. An engineered enzyme that targets circulating lactate to alleviate intracellular NADH:NAD(+) imbalance. Nat. Biotechnol. 2020, 38, 309–313.
- Ogiwara, H.; Takahashi, K.; Sasaki, M.; Kuroda, T.; Yoshida, H.; Watanabe, R.; Maruyama, A.; Makinoshima, H.; Chiwaki, F.; Sasaki, H.; et al. Targeting the Vulnerability of Glutathione Metabolism in ARID1A-Deficient Cancers. Cancer Cell 2019, 35, 177–190.e178.
- Zhang, J.; Li, X.; Han, X.; Liu, R.; Fang, J. Targeting the Thioredoxin System for Cancer Therapy. Trends Pharmacol. Sci. 2017, 38, 794–808.
- Costa, T.J.; Barros, P.R.; Arce, C.; Santos, J.D.; da Silva-Neto, J.; Egea, G.; Dantas, A.P.; Tostes, R.C.; Jimenez-Altayo, F. The homeostatic role of hydrogen peroxide, superoxide anion and nitric oxide in the vasculature. Free Radic. Biol. Med. 2021, 162, 615–635.
- Reddi, A.R.; Culotta, V.C. SOD1 integrates signals from oxygen and glucose to repress respiration. Cell 2013, 152, 224–235.
- Montllor-Albalate, C.; Colin, A.E.; Chandrasekharan, B.; Bolaji, N.; Andersen, J.L.; Wayne Outten, F.; Reddi, A.R. Extra-mitochondrial Cu/Zn superoxide dismutase (Sod1) is dispensable for protection against oxidative stress but mediates peroxide signaling in Saccharomyces cerevisiae. Redox Biol. 2019, 21, 101064.
- Yang, M.; Cobine, P.A.; Molik, S.; Naranuntarat, A.; Lill, R.; Winge, D.R.; Culotta, V.C. The effects of mitochondrial iron homeostasis on cofactor specificity of superoxide dismutase 2. EMBO J. 2006, 25, 1775–1783.
- Mohammedi, K.; Bellili-Munoz, N.; Marklund, S.L.; Driss, F.; Le Nagard, H.; Patente, T.A.; Fumeron, F.; Roussel, R.; Hadjadj, S.; Marre, M.; et al. Plasma extracellular superoxide dismutase concentration, allelic variations in the SOD3 gene and risk of myocardial infarction and all-cause mortality in people with type 1 and type 2 diabetes. Cardiovasc. Diabetol. 2015, 14, 845.
- Dong, S.; Dong, Y.; Liu, B.; Liu, J.; Liu, S.; Zhao, Z.; Li, W.; Tian, B.; Zhao, R.; He, F.; et al. Guiding Transition Metal-Doped Hollow Cerium Tandem Nanozymes with Elaborately Regulated Multi-Enzymatic Activities for Intensive Chemodynamic Therapy. Adv. Mater. 2022, 34, e2107054.
- Robbins, M.E.; Cho, H.Y.; Hansen, J.M.; Luchsinger, J.R.; Locy, M.L.; Velten, M.; Kleeberger, S.R.; Rogers, L.K.; Tipple, T.E. Glutathione reductase deficiency alters lung development and hyperoxic responses in neonatal mice. Redox Biol. 2021, 38, 101797.
- Luzzatto, L.; Arese, P. Favism and Glucose-6-Phosphate Dehydrogenase Deficiency. N. Engl. J. Med. 2018, 378, 60–71.
- Harel, M.; Aharoni, A.; Gaidukov, L.; Brumshtein, B.; Khersonsky, O.; Meged, R.; Dvir, H.; Ravelli, R.B.; McCarthy, A.; Toker, L.; et al. Structure and evolution of the serum paraoxonase family of detoxifying and anti-atherosclerotic enzymes. Nat. Struct. Mol. Biol. 2004, 11, 412–419.
- Fumarola, S.; Cecati, M.; Sartini, D.; Ferretti, G.; Milanese, G.; Galosi, A.B.; Pozzi, V.; Campagna, R.; Morresi, C.; Emanuelli, M.; et al. Bladder Cancer Chemosensitivity is Affected by Paraoxonase-2 Expression. Antioxidants 2020, 9, 175.
- Campagna, R.; Bacchetti, T.; Salvolini, E.; Pozzi, V.; Molinelli, E.; Brisigotti, V.; Sartini, D.; Campanati, A.; Ferretti, G.; Offidani, A.; et al. Paraoxonase-2 Silencing Enhances Sensitivity of A375 Melanoma Cells to Treatment with Cisplatin. Antioxidants 2020, 9, 1238.
- Devarajan, A.; Bourquard, N.; Hama, S.; Navab, M.; Grijalva, V.R.; Morvardi, S.; Clarke, C.F.; Vergnes, L.; Reue, K.; Teiber, J.F.; et al. Paraoxonase 2 deficiency alters mitochondrial function and exacerbates the development of atherosclerosis. Antioxid. Redox Signal. 2011, 14, 341–351.
- Guan, T.; Song, J.; Wang, Y.; Guo, L.; Yuan, L.; Zhao, Y.; Gao, Y.; Lin, L.; Wang, Y.; Wei, J. Expression and characterization of recombinant bifunctional enzymes with glutathione peroxidase and superoxide dismutase activities. Free Radic. Biol. Med. 2017, 110, 188–195.
- Cao, L.; Wu, G.; Zhu, J.; Tan, Z.; Shi, D.; Wu, X.; Tang, M.; Li, Z.; Hu, Y.; Zhang, S.; et al. Genotoxic stress-triggered beta-catenin/JDP2/PRMT5 complex facilitates reestablishing glutathione homeostasis. Nat. Commun. 2019, 10, 3761.
- Floros, K.V.; Cai, J.; Jacob, S.; Kurupi, R.; Fairchild, C.K.; Shende, M.; Coon, C.M.; Powell, K.M.; Belvin, B.R.; Hu, B.; et al. MYCN-Amplified Neuroblastoma Is Addicted to Iron and Vulnerable to Inhibition of the System Xc-/Glutathione Axis. Cancer Res. 2021, 81, 1896–1908.
- Schaupp, C.M.; Botta, D.; White, C.C.; Scoville, D.K.; Srinouanprachanh, S.; Bammler, T.K.; MacDonald, J.; Kavanagh, T.J. Persistence of improved glucose homeostasis in Gclm null mice with age and cadmium treatment. Redox Biol. 2022, 49, 102213.
- Kelly-Aubert, M.; Trudel, S.; Fritsch, J.; Nguyen-Khoa, T.; Baudouin-Legros, M.; Moriceau, S.; Jeanson, L.; Djouadi, F.; Matar, C.; Conti, M.; et al. GSH monoethyl ester rescues mitochondrial defects in cystic fibrosis models. Hum. Mol. Genet. 2011, 20, 2745–2759.
- Navarro, J.; Obrador, E.; Carretero, J.; Petschen, I.; Avino, J.; Perez, P.; Estrela, J.M. Changes in glutathione status and the antioxidant system in blood and in cancer cells associate with tumour growth in vivo. Free Radic. Biol. Med. 1999, 26, 410–418.
- McCarver, A.C.; Lessner, D.J. Molecular characterization of the thioredoxin system from Methanosarcina acetivorans. FEBS J. 2014, 281, 4598–4611.
- Hanschmann, E.M.; Petry, S.F.; Eitner, S.; Maresch, C.C.; Lingwal, N.; Lillig, C.H.; Linn, T. Paracrine regulation and improvement of beta-cell function by thioredoxin. Redox Biol. 2020, 34, 101570.
- Peskin, A.V.; Meotti, F.C.; Kean, K.M.; Gobl, C.; Peixoto, A.S.; Pace, P.E.; Horne, C.R.; Heath, S.G.; Crowther, J.M.; Dobson, R.C.J.; et al. Modifying the resolving cysteine affects the structure and hydrogen peroxide reactivity of peroxiredoxin 2. J. Biol. Chem. 2021, 296, 100494.
- Perez-Jimenez, R.; Li, J.; Kosuri, P.; Sanchez-Romero, I.; Wiita, A.P.; Rodriguez-Larrea, D.; Chueca, A.; Holmgren, A.; Miranda-Vizuete, A.; Becker, K.; et al. Diversity of chemical mechanisms in thioredoxin catalysis revealed by single-molecule force spectroscopy. Nat. Struct. Mol. Biol. 2009, 16, 890–896.
More