Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Shian-Ren Lin and Version 2 by Peter Tang.
Prostate cancer (PCa) is one of the most prevalent and malignant cancer types in men, which causes more than three-hundred thousand cancer death each year. At late stage of PCa progression, bone marrow is the most often metastatic site that constitutes almost 70% of metastatic cases of the PCa population.
- Wnt
- Ras
- prostate cancer
- bone metastasis
1. Introduction: Metastasis of Prostate Cancer (PCa)
Bone marrow is the most frequent metastatic site of PCa, which include 72.8% of all metastatic cases, at over three times higher than the second most frequent site [1]. Knowing the underlying mechanism of osteophilic nature in PCa metastasis is urgent for monitoring and preventing PCa bone metastasis. Interestingly, PCa metastasis could be divided into two types: osteoblastic and osteoclastic, and that is apparently triggered by different mechanism [2]. Which molecules or cells are key factors in choosing the bone metastasis type? These two questions have attracted the attention of researchers in cancer biology, in the hope that they can be applied to an early diagnosis of cancer metastasis.
2. Mechanism of BM in PCa
2.1. Epithelial-Mesenchymal Transition (EMT) and Tumor Invasion
2.1.1. TGF-β/MAPK-Mediated EMT
During metastasis, PCa cells transform into mesenchymal-like cells, which helps cells to invade through the basal lamina and survive in the blood circulation without cell-cell interactions [3][9]. The EMT in PCa is activated by four mediators of snail, slug, twist, and Zinc finger E-box-binding homeobox 1 (ZEB1), while four signaling pathways play dominant roles here: the transforming growth factor (TGF) receptor (TGFR)/mothers against decapentaplegic homolog (Smads)/mitogen-activated protein kinase (MAPK) signaling cascade, nuclear factor (NF)-κB signaling, and the receptor tyrosine kinase (RTK) cascade [3][9]. Wa et al. reported that MAPK can also promote the EMT through inhibiting Rb phosphorylation, which is one of the main suppressors in cell cycle regulation [4][10]. Other signaling factors regulate the TGF-β-MAPK axis in PCa BM. Lue et al. described how MAPK is upregulated by heparin-binding epidermal growth factor (HB-EGF)-mediated signaling, in which the HB-EGF is cut from the PCa surface by the zinc transporter, ZIP6 (LIV-1), which stimulates matrix metalloproteinase (MMP)-2/9 upregulation in other PCa cells [5][11]. Interferon (INF)-induced transmembrane protein 3 (IFITM3), a highly expressed transmembrane protein in cancer cells, is believed to be associated with metastasis in various cancers, e.g., acute myeloid leukemia, hepatomas, and gliomas [6][7][8][9][12,13,14,15]. In PCa, IFITM3 promotes fibroblast growth factor (FGF) expression and promotes TGF-β production, which elicits the EMT in neighboring cells in a paracrine manner [10][16]. On the contrary, the TGF-β-mediated EMT can be retarded via microRNA (miR) regulation. miR-33a-5p reduces TGFR 1 expression, which affects its offset by increasing the ZEB1 copy number [11][17]. Moreover, the TGFR and Smad2/4 are suppressed by miR-505-3p and miR-19a-3p [4][12][10,18]. Those studies clearly depicted a regulatory network in TGF-β-mediated BM in PCa cells.2.1.2. NF-κB Activation after Androgen Receptor (AR) Signaling Deprivation
NF-κB signaling pushes cancer metastasis in multiple directions, such as stimulating MMP expressions and regulating cell adhesion molecules, according to previous studies [13][19]. The tumor necrosis factor (TNF)-α receptor (TNFR) promotes inhibitor of NF-κB (IκB) kinase (IKK) activity, which blocks the binding of IκB to NF-κB and releasing the active form of NF-κB [14][20]. Active NF-κB ultimately triggers hypoxia-inducible factor (HIF)-1α expression and subsequently induces the EMT [15][21]. In addition to TNFR signaling, NF-κB can also be activated by TNF-related weak inducer of apoptosis (TWEAK)/TNFR superfamily member 12A (TNFRSF12A, also known as Fn14)-mediated IKK-β activation and downregulation of miR-210-3p-triggered suppressor of cytokine signaling 1 (SOCS1) and TNFAIP3-interacting protein 1 (TNIP1) [16][17][22,23]. Conversely, activated AR and its cofactor FOXA1 inhibits TWEAK/Fn14/IKK-β activation through directly binding to an androgen-binding element in TWEAK and the Fn14 promoter/enhancer in order to reduce TWEAK and Fn14 transcription [16][22]. After androgen deprivation therapy (ADT), some castration-sensitive PCa cells will transit into CRPC cells, which is the beginning of PCa metastasis [18][19][24,25]. Izumi and Mizokami summarized the characteristic of C-C motif ligand 5 (CCL5) in regulating AR expression, in which CCL5 downregulates AR expression [20][26]. The above studies not only evaluated the second central signaling axis in PCa BM, but also evaluated how CRPC is induced.2.1.3. Contribution of PI3K/Akt/MAPK Signaling in EMT of PCa
The third signaling pathway that is involved in PCa BM is the phosphoinositide 3-kinase (PI3K)/Akt signaling cascade, which originates from the activation of the epidermal growth factor (EGF) and vascular endothelial growth factor (VEGF). In general, the activation of EGF and VEGF receptors (EGFR and VEGFR) stimulates the Ras/Raf/MAPK kinase (MEK)/MAPK signaling cascade, which is involved in tumor progression or the PI3K/Akt/mammalian target of rapamycin (mTOR) cascade that promotes cell growth and the EMT [21][22][27,28]. In PCa, EGF signaling accompanies alterations in miR-96 and miR-30 expression, which act contrary to each other. EGF signaling promotes miR-96 expression, which attends to the degradation of E26 transformation-specific variant 6 (ETV6, also known as TEL, a transcriptional repressor in regulating embryonic and hematopoietic cell proliferation) that blocks the expression of the TWIST1 oncogene [23][24][25][26][29,30,31,32]. Kao et al. reported that EGF signaling inhibits miR-30 expression, which directly reduces ETS-related gene (ERG) expressions [27][33]. In addition to EGF signaling, miR-30 can also be reduced by Src/STAT3, which is mediated by the VEGFR/NRP-1/c-Met/Mcl-1 cascade [27][28][33,34]. When tracing upstream of VEGF signaling in PCa metastasis, reprogramming of glucose metabolism was identified as a critical step for the EMT [29][35]. The core regulator of glucose metabolism, AMP-activated protein kinase (AMPK), triggers cell migration-inducing protein (CEMIP) overexpression through the AMPK/glycogen synthase kinase 3β (GSK3β)/β-catenin cascade for which CEMIP mediates VEGF and MMP-2 upregulation and subsequently results in anoikis resistance [30][36]. In addition to AMPK, VEGF expression can also be modulated by HIF-1α. The RTK signaling cascade promotes mTOR phosphorylation, which elevates HIF-1α expression [31][37]. Furthermore, HIF-1α triggers pyruvate kinase M2 (PKM2) as a transcription factor that stimulates neuroendocrine markers, like oct4 and VEGF [32][33][38,39]. The EMT can be activated by PI3K/Akt- and MAPK-mediated mTOR activation, which promotes EMT and metastasis through the phosphorylation of eukaryotic translation initiation factor 4E-binding protein 1 (EIF4EBP1) [34][35][36][40,41,42]. Bi et al. and Tang et al. demonstrated that miR-153 and miR-133a-3p are involved in PCa BM, in which miR-153 exacerbates the EMT through inhibiting phosphatase and tensin homolog (PTEN), and miR-133a-3p acts inversely through reducing growth factor receptor expressions [35][37][41,43]. Those studies provided further insights into RTK signaling in the EMT, and not just in maintaining cell survival [38][44].2.1.4. Other Minor EMT contributors
Other minor mediators that are discovered to be associated with PCa BM include KDM8, miR-145, and CCCTC-binding factor (CTCF). In the previous paragraph, we discussed the inhibitory characteristics of the AR in PCa metastasis were discussedis. However, KDM8, an AR transcriptional enhancer, which can accompany with AR binding to the enhancer and promoter region of ATPase family AAA domain-containing protein 2 (ANCCA) and enhancer of zeste homolog 2 (EZH2) gene, results in PCa metastasis, neuroendocrine differentiation, and growth promotion [39][45]. miR-145 is a tumor-suppressing microRNA in various cancers, including PCa, head and neck squamous cell carcinoma, and breast cancer [40][46]. Reports from Guo et al., Ren et al., and Huang et al. showed that miR-145 is activated by p53 and it inhibits mesenchymal and cancer stem cell markers, like fibronectin, vimentin, cluster of differentiation 44 (CD44), and c-Myc via inhibiting two mediators: human enhancer of filamentation 1 (hEF1) and Krüppel-like factor 4 (KLF4) [41][42][43][47,48,49]. However, the AR and KLF4 form reciprocal feedback for promoting expression and that also results in the inhibition of PCa proliferation and metastasis [44][50]. KLF4 is a crucial regulator of normal cell proliferation and it can be a tumor suppressor or an oncogene, depending on the tissue, tumor type, or cancer stage [45][46][47][51,52,53]. miR-1 acts as a tumor suppressor that is involved in regulating BM, and miR-1 is tightly regulated by the AR [48][54]. Siu et al. demonstrated that KLF4 functions as a transcription factor to activate the AR-miR-1 signaling pathway to constrain the tumor-suppressive role of miR-1 [44][50]. CTCF, a downstream transcription activator of Smad and Myc signaling, contributes to slowing down metastasis in PCa through directly interacting with the miR-127-3p promoter, in which the downregulation of miR-127-3p accompanies proteasome subunit beta type-5 (PSMB5) elevation and subsequently leads to the activation of BM [49][50][51][55,56,57]. All of the above research draws a large picture of the regulatory mechanism of BM in PCa cells, and Figure 1 summarizes these signaling cascades. In the next section, we discuss how PCa cells interact with bone stromal cells and the function of PCa/osteocytes in BM.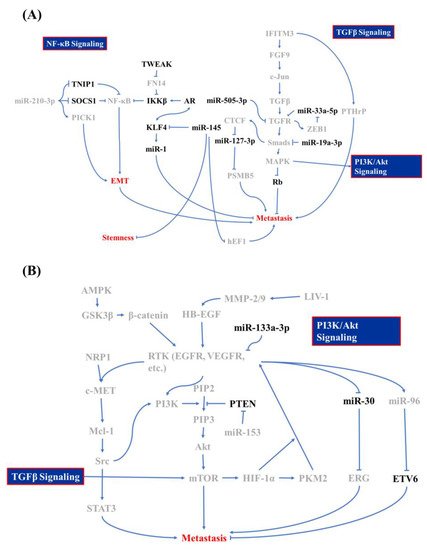
Figure 1. Molecular mechanism of bone metastasis in prostate cancer. Signaling regulators of (A) the nuclear factor (NF)-κB, transforming growth factor (TGF)-β, and (B) phosphatidylinositol-3-kinase (PI3K)/Akt signaling cascade whose is involved in bone metastasis of prostate cancer and which acts as promoters are labeled in gray and inhibitors are in black.