Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 3 by Jing Xu and Version 2 by Jing Xu.
Maple Syrup Urine Disease (MSUD) is caused by a deficiency of branched-chain α-ketoacid dehydrogenase (BCKD). It is a metabolic disorder characterized by increased levels of branched-chain amino acids (BCAAs) and their respective branched-chain α-ketoacids (BCKAs) [1].
- Maple Syrup Urine Disease
- branched-chain amino acids
- Metabolism
- Neurological disorder
1. Branched-Chain Amino Acid Metabolism and Maple Syrup Urine Disease
Branched-chain α-ketoacid dehydrogenase deficiency, commonly known as Maple Syrup Urine Disease (MSUD), is a metabolic disorder characterized by increased levels of branched-chain amino acids (BCAAs) and their respective branched-chain α-ketoacids (BCKAs) [1] (Figure 1). The three branched-chain amino acids leucine, isoleucine, and valine are unable to be synthesized by animals, and their metabolism is essential for protein synthesis and cell signaling [2]. Therefore, when BCAA metabolism is disrupted, such as in MSUD, a variety of pathologic changes arise.
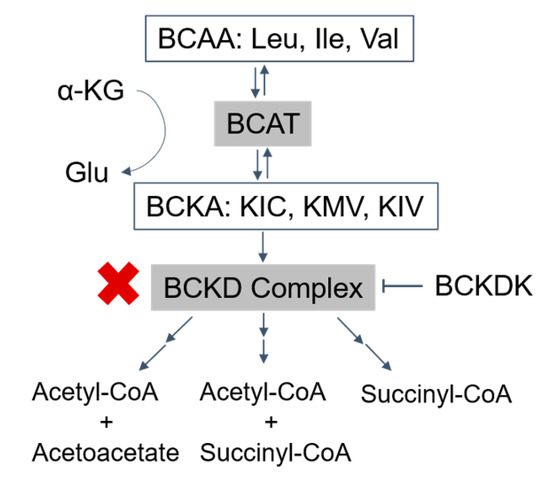
Figure 1. Catabolism pathways of branched-chain amino acids (BCAAs): the BCAAs are transmitted by branched-chain amino acid transaminase (BCAT) to form branched-chain α-ketoacids (BCKAs) (α-ketoisocaproate (KIC), α-keto-β-methylvalerate (KMV), and α-ketoisovalerate (KIV)), which are irreversibly oxidized by the BCKD complex. The final products, acetyl-CoA, acetoacetate, and succinyl-CoA, are produced after a series of further reactions (→→). BCKDK phosphorylates and inhibits the activity of the BCKD complex (T arrow). The activity of the BCKD complex is decreased in maple syrup urine disease (red ‘X’). BCAA, branched-chain amino acids; Leu, leucine; Ile, isoleucine; Val, valine; α-KG, α-ketoglutarate; Glu, glutamate; BCAT, branched-chain amino acid transaminase; BCKAs, branched-chain α-ketoacids; KIC, α-ketoisocaproate; KMV, α-keto-β-methylvalerate; KIV, α-ketoisovalerate; BCKDK, branched-chain ketoacid dehydrogenase kinase.
The prevalence of MSUD in the United States is estimated to be 1:200,000, though some populations have much higher incidence rates. Such populations include the Mennonite population, with a reported incidence as high as nearly 1:350; the Galician population in Spain, with an incidence of 1:52,500; and the Ashkenazi Jewish population [3].
Normally, the first step in BCAA metabolism is transamination by branched-chain amino acid transaminases (BCATs) to form branched-chain α-ketoacids (BCKAs), including α-ketoisocaproate (KIC), α-keto-β-methylvalerate (KMV), and α-ketoisovalerate (KIV) for leucine, isoleucine, and valine, respectively (Figure 1). In the next rate-limiting step of the pathway, the branched-chain ketoacid dehydrogenase complex (BCKDC) catalyzes the oxidative decarboxylation of the α-ketoacids.
MSUD arises from biallelic loss of function mutations in one of the genes that encode BCKDC subunits. Decreased BCKDC activity results in the failure of BCKAs to be oxidized into their respective end products, leading to an accumulation of BCAAs and BCKAs [5]. Due to BCAA and BCKA elevations, patients with MSUD can demonstrate acute severe ketoacidosis and neurological symptoms such as apnea, seizures, and coma as well as chronic features such as poor feeding, ataxia, motor delay, and intellectual disability due to amino acid and neurotransmitter imbalances [5,6].
Current therapies for MSUD include dietary therapy and liver transplantation. Dietary therapy requires restriction of BCAA by limiting protein in the diet and consumption of medical formulas. However, it is unclear how diet therapy affects the biochemistry of the CNS. Patients treated in this manner may still manifest a high burden of neuropsychological symptoms [5].
Liver transplantation aims to replace functional BCKDCs in the liver and to thus promote BCAA metabolism in peripheral tissue [7]. A recent study demonstrated that transplants can restore homeostasis and may arrest neurocognitive effects [5]. Unfortunately, liver transplantation was not found to improve preexisting impairments and patients are susceptible to postoperative complications and require long-term immunosuppression [7]. One long-term follow-up study of MSUD patients that had undergone treatment found that 40% of these transplantation patients have required management for acute rejection [2]. Additionally, there is evidence of similar neuropsychiatric morbidity in MSUD patients who had and had not undergone liver transplantation [8].
Recent studies have shown evidence of dysregulation of certain amino and organic acids in the CNS of MSUD patients, related to the disruption of BCAAs catabolism in these individuals. One study found decreased levels of glutamate and N-acetyl-aspartate (NAA) in the brain of MSUD patients [9], while another found decreased levels of phenylalanine, tryptophan, methionine, and tyrosine [5] and others found evidence of elevated lactate [10,11,12,13,14].
One proposed mechanism for the decrease in glutamate and elevation in lactate levels is that, in MSUD patients, the increased levels of BCKAs (specifically α-ketoisocaproic acid) leads to reversed flux through BCAT, which normally catalyzes the conversion of BCAAs and α-ketoglutarate to BCKAs and glutamate. As a result, this reversal leads to decreased glutamate levels and increased levels of BCAAs (specifically leucine) and α-ketoglutarate [8] (Figure 2a). The excess α-ketoglutarate, along with aspartate, can then converted via aspartate aminotransferase to form oxaloacetate and glutamate [15] (Figure 2b), depleting brain aspartate levels. This excess α-ketoglutarate is also thought to drive the formation of pyruvate (a precursor to lactate) from alanine and α-ketoglutarate (Figure 2c). It was argued, though, that glutamate can be produced by the transaminase reaction from aspartate and alanine (Figure 2b,c), compensating for the reduced amount of glutamate. Another explanation for the altered levels of glutamate and lactate involves the anaplerotic role of valine and isoleucine. Valine and isoleucine significantly refill the tricarboxylic acid (TCA) cycles via succinyl-CoA. As a result, other intermediates in the TCA cycle are depleted over the time. This leads to a reduced ability of α-ketoglutarate to produce glutamate and an increased reliance on anaerobic glycolysis that produces lactate.
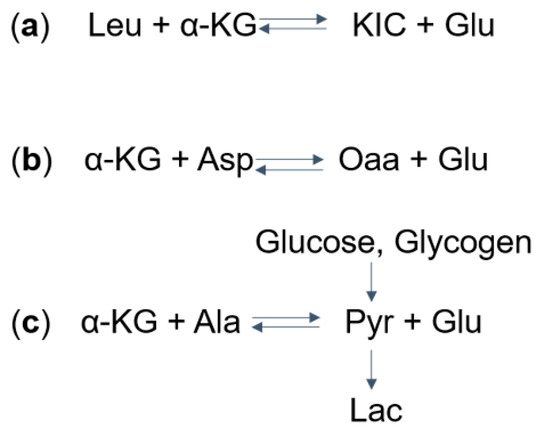
Figure 2. Mechanisms of (a) Glu, (b) Asp, and (c) Lac dysregulation in Maple syrup urine disease (MSUD): (a) take Leu as an example, the aminotransferase catalyzes the reversible reaction to form α-KIC and Glu from leucine and α-KG. The excess α-KIC in MSUD drives the reaction in reverse, depletes Glu, and generates α-KG. (b) The increased α-KG then drives the transaminase reaction to deplete Asp and to form Oaa and Glu. (c) Also, the α-KG can deplete Ala and produce Glu and Pyr. Pyr, which can also be formed from glucose and glycogen, is further converted into Lac. MSUD, maple syrup urine disease; Leu, leucine; α-KG, α-ketoglutarate; KIC, α-ketoisocaproate; Glu, glutamate; Asp. Aspartate; Oaa, oxaloacetate; Ala, alanine; Pyr, pyruvate; Lac, lactate.
There is also evidence of decreased phenylalanine, tyrosine, tryptophan, and methionine in the brain of MSUD patients [5]. This is thought to arise from the excess levels of BCAAs (specifically leucine) in such patients. Leucine competes with these other amino acids for entry into the brain via the large neutral amino acid transporter (LAT1, encoded by SLC7A5). As leucine has the highest affinity for the transporter, excessive levels in MSUD patients reduce the ability for these other amino acids to enter the brain [8,9].
Overall, the dysregulation of these amino acids may lead to brain dysfunction, predisposing MSUD patients to cognitive and psychiatric disabilities despite major clinical interventions such as liver transplant [5]. This review will therefore discuss the potential impacts of these dysregulated AAs in the brain of MSUD patients.