Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Toshihiro Ihara and Version 2 by Catherine Yang.
Various DNA structures, including specific metal ion complexes, have been designed based on the knowledge of canonical base pairing as well as general coordination chemistry. The role of metal ions in these studies is quite broad and diverse. Metal ions can be targets themselves in analytical applications, essential building blocks of certain DNA structures that one wishes to construct, or they can be responsible for signal generation, such as luminescence or redox.
- DNA conjugate
- metal ion
- triple helix
- silver ion
- lanthanide
1. Stabilization of a Parallel-Motif DNA Triplex by Silver Ion
When designing the ligands for specific sequences in DNA duplexes, triple helix formation is a useful recognition motif, inasmuch as the formation of the base triplet follows the simple rule of complementary Hoogsteen hydrogen bonding, CG.C+ and TA.T, for the parallel motif of the triplex. However, the triplexes containing CG.C+ triplets form only in a weak acidic solution, because the N3 position of cytosines (pKa = 4.5) in the third strand must be protonated to fulfill its complementarity [1][18]. With the aim of achieving sufficient stability under physiological conditions, a large quantity of chemically modified DNA has been developed by taking advantage of the highly advanced techniques of organic synthesis [2][19].
The silver ion was expected to displace an N3 proton of a cytosine in the CG.C+ to form a metal ion-mediated base triplet, CG.CAg+, as shown in Figure 1a. This process was expected to stabilize parallel motif triplexes even at neutral pH.
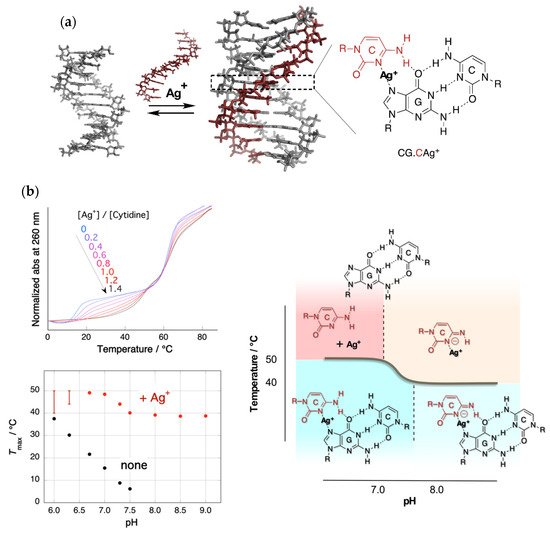
Figure 1. Triplex stabilization by silver ion. (a) Structure of the triplex and CG.CAg+ base triplet; (b) upper left: UV melting curves in the presence of Ag+ with various feeding ratios. Only the temperature of the triplex–duplex transition increased with the addition of Ag+. Bottom left: pH dependence of the melting temperatures of triplex in the absence and presence of Ag+. The melting temperature in the presence of Ag+ consists of two pH-independent regions. Right: Phase diagram of the structure of Ag+-mediated nucleobase complexes.
Figure 1b shows the UV melting curves at a pH of 7.0 and pH dependence of the temperatures of triplex–duplex transition. Surprisingly, the addition of an equal amount of Ag+ (to CG.C+) increased the melting temperature of the triplex by more than 30 °C under neutral conditions [3][20]. In the absence of Ag+, the relation of the melting temperature to pH was clearly evident. Meanwhile, in the presence of Ag+, the correlation disappeared, and a biphasic feature consisting of two temperature-independent regions was observed.
2. Metal Ion-Directed Dynamic Splicing of DNA through Global Conformational Change by Intramolecular Complexation
The metal ion-directed global conformational control of DNA was performed as follows. Two terpyridine units were built into the distal sites on the DNA backbone to prepare a conjugate, i.e., terpy2DNA. The two terpyridines formed a stable intramolecular 1:2 complex, [M(terpy)2]2+, with divalent transition metal ions, M2+, namely Fe2+, Ni2+, Cu2+, and Zn2+. By the specific formation of an intramolecular metal complex, a part of the sequence of the DNA in between the two terpyridine units was reversibly excluded, and the two flanking external DNA segments were directly connected with each other to form an Ω-shaped structure presenting a new sequence (Figure 2). This can be regarded as a metal ion-directed reversible edition of the DNA sequence or dynamic DNA splicing [4][26].
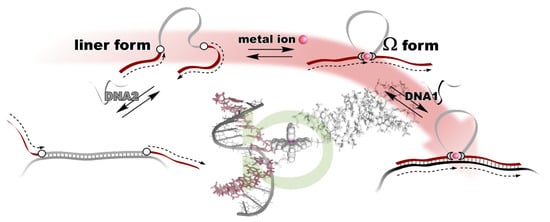
Figure 2. Metal ion-directed reversible edition of the DNA sequence. The sequence of terpy2DNA is edited by intramolecular complexation with appropriate metal ions through Ω-shaped global conformational change.
Conformational control of terpy2DNA was confirmed via UV melting with the complementary tandem sequence of the two external segments. The results show that the duplex structure was significantly stabilized in the presence of an equimolar amount (to terpy2DNA) of transition metal ions. In addition, in the presence of the metal ions, the shape of the melting curves changed to be more cooperative, indicating that the two sequences outside the terpyridines were cooperatively dissociated in a narrow temperature range. The dependences of duplex stabilization on the metal ion feeding ratio (r = [M2+]/[terpy2DNA]) were different for each of the metal ions. In the case of Fe2+ and Ni2+, the duplex remained stable even when additional metal ions were added to r = 2 or 3. In contrast, the duplex was destabilized at higher feeding ratios of Cu2+ and Zn2+. The stability of the duplex was maintained even in the presence of the excess amounts of Fe2+ and Ni2+, because the Ω-shaped conformation of terpy2DNA was preserved due to the magnitudes of the two successive binding constants with terpyridine, K1 < K2. As for Cu2+ and Zn2+, the global conformation of terpy2DNA was changed from Ω-form to a linear form accompanying the transition of the complex types formed on terpy2DNA from [M(terpy)2]2+ (on terpy2DNA∙M2+) to 2[M(terpy)]2+ (on terpy2DNA∙2M2+) with increasing amounts of ions due to their binding properties with terpyridine, K1 > K2. This indicates that the general trend of the complexation of transition metal ions found in the text books of coordination chemistry is still valid on DNA.
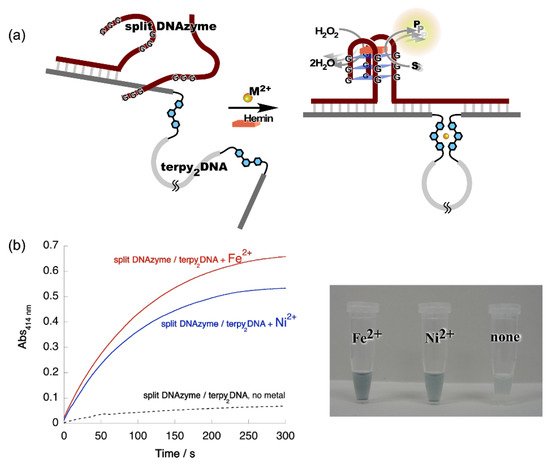
Figure 3. Metal ion-directed regulation of DNAzyme activity. (a) Allosteric regulation of split DNAzyme activity by metal ion-directed dynamic sequence edition of the template, terpy2DNA. (b) Left: Time courses of the ABTS oxidation by the split DNAzyme with terpy2DNA in the presence of Fe2+ and Ni2+. Red, split DNAzyme/terpy2DNA + Fe2+; blue, split DNAzyme/terpy2DNA + Ni2+; black, split DNAzyme/terpy2DNA, no metal ions. Right: Images of reaction solutions shown in the time courses.
3. Reconstruction of Luminescent Lanthanide Complexes on DNA and Their Analytical Applications
The sequences of these split probes were designed so as to form a tandem duplex with targets (templates) with their auxiliary units facing each other, providing a microenvironment to accommodate Ln3+ (Figure 4a) [5][27]. The results of time-resolved luminescence studies showed that the formation of luminous ternary complexes, EDTA/Ln3+/phen, depends on the sequence of the targets. The intensity of the luminescence is affected by the binding affinities of the probes or the local structural disruption caused by one-base mispairing [6][28].
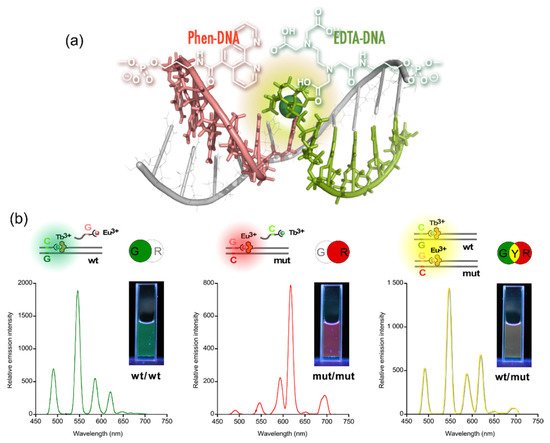
Figure 4. Multicolored allele typing using time-resolved luminescence from lanthanide complexes (Tb3+ and Eu3+) cooperatively formed with a pair of split probes. (a) The structure of the Ln3+ complex formed on tandem duplex of the split probes with target sequence. (b) Allele typing of thiopurine S-methyltransferase gene.
This technique was applied to the multicolored allele typing based on single nucleotide polymorphisms (SNPs) in thiopurine S-methyltransferase gene by the concomitant use of the two capture probes, which are complementary to a part of the wild-type (wt) and the mutant (mut) of the gene. First, the capture probes for wt and mut were mixed with equimolar amounts of Tb3+ and Eu3+, respectively. Both the allele-specific capture probe with Ln3+ and the sensitizer probe were then added to three different solutions containing the targets, wt/wt, mut/mut, and wt/mut. The solutions emitted distinctive colors, i.e., green, red, and yellow for wt/wt, mut/mut, and wt/mut, respectively; the colors were identifiable with the naked eye (Figure 4b) [7][29].
The technique of binary luminous Ln complex formation was applied as ATP sensor using the platform of molecular beacon (LCMB). The ATP aptamer (iATP) was used as an interface for the application of LCMB to ATP sensing. The sequence of LCMB was designed to be complementary to a part of iATP (Figure 5a). Scarce emission was observed in the presence of the DNA complementary to the loop region (iATP); the auxiliary units were separated from each other when the duplex was formed (“off” state). With the addition of ATP to the LCMB/iATP duplex, the fluorescence signal turned on as the result of the restoration of LCMB stem-loop structure accompanying the displacement of iATP from LCMB by ATP. A highly specific response was observed for ATP among NTPs, as shown in Figure 5b [8][30].
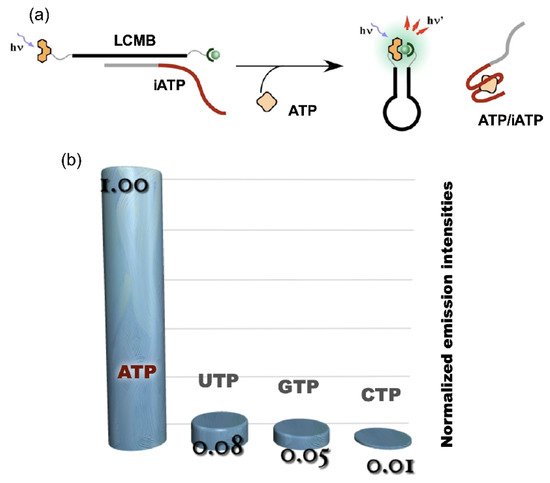
Figure 5. ATP sensing using LCMB and iATP. (a) operating principle of ATP sensing using competitive reaction over iATP between ATP and LCMB; (b) luminescence signal response of ATP sensor to NTPs.
Nonenzymatic amplification of the luminescent signal from the Ln complexes on the DNA scaffold was performed through catalytic hairpin assembly (CHA) and hybridization chain reaction (HCR), which are the typical DNA circuits consisting of the autonomous successive strand exchange reactions [9][10][31,32]. Figure 6a shows the scheme of the HCR amplification. The luminescence signal significantly increased with the progress of HCR after target addition. Signal contrast was very high, and the sequence selectivity was preserved in this system [10][32]. To improve the amplification rate, the system was redesigned to form a cruciform product consisting of four hairpins by catalytic hairpin assembly (CHA) (Figure 6b).
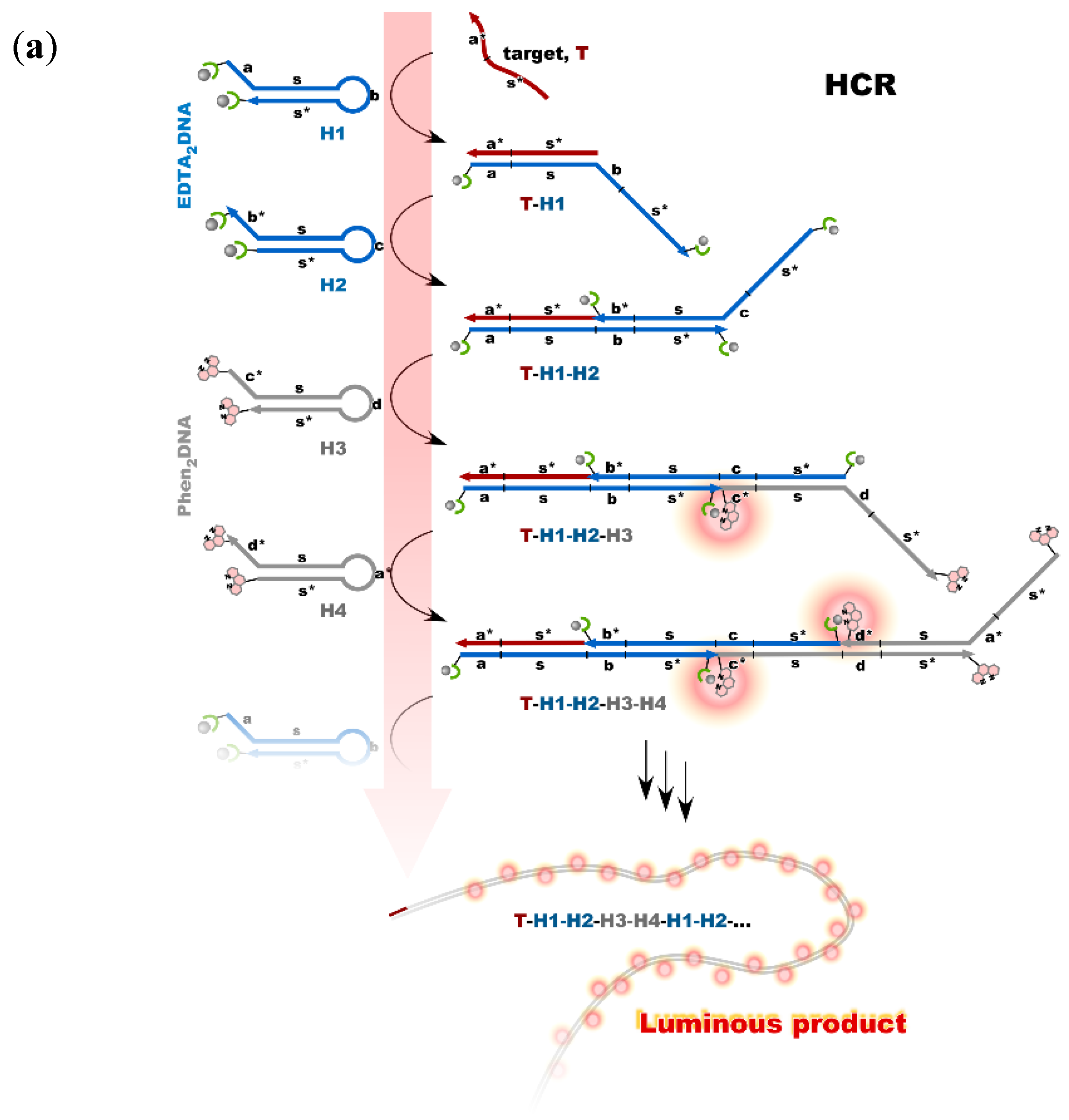
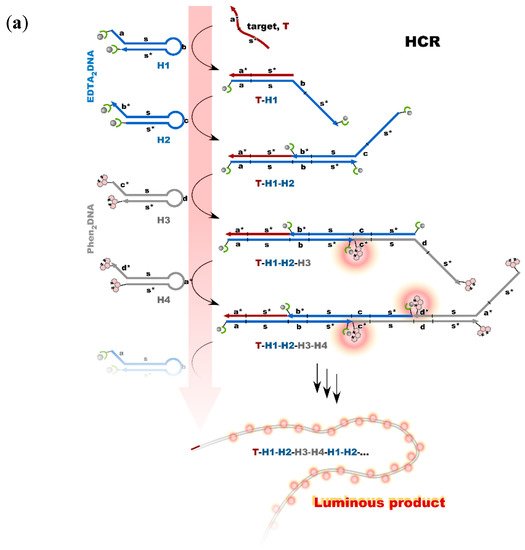
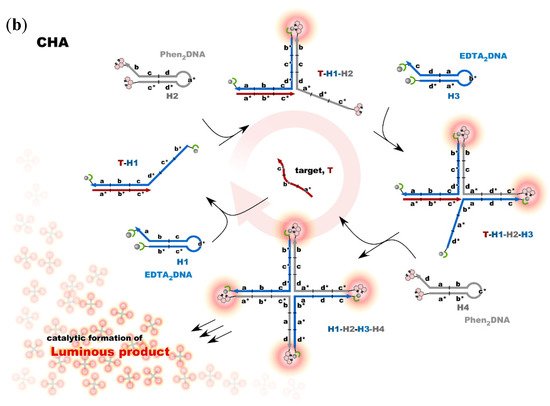
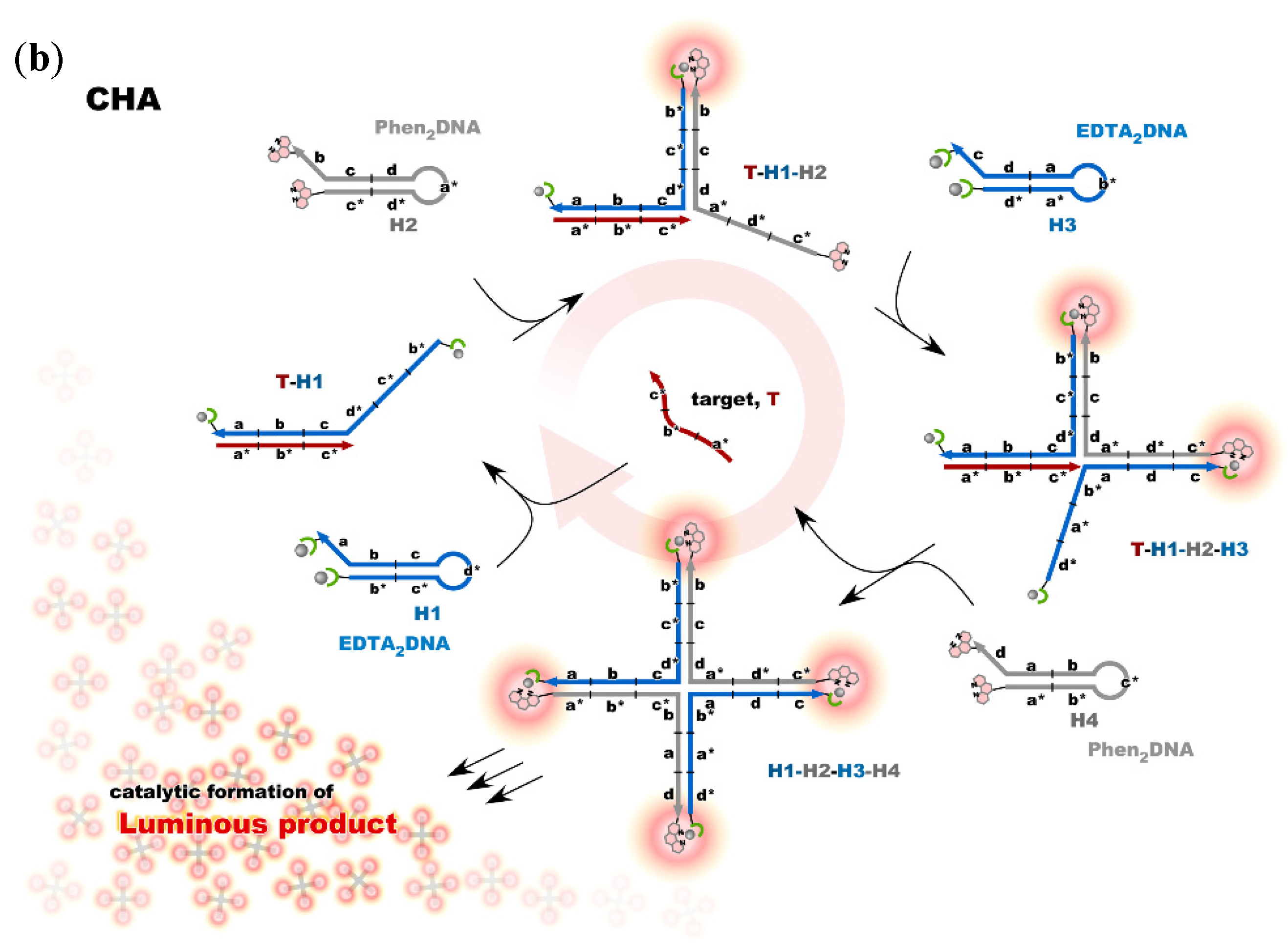
Figure 6. Nonenzymatic signal amplification by DNA circuits: (a) Luminous DNA wire was produced by target-initiated HCR; (b) Luminous cruciform DNA was produced by CHA.