Renal inflammation is an initial pathological process during progressive renal injury regardless of the initial cause. Macrophage migration inhibitory factor (MIF) is a truly proinflammatory stress mediator that is highly expressed in a variety of both inflammatory cells and intrinsic kidney cells. MIF is released from the diseased kidney immediately upon stimulation to trigger renal inflammation by activating macrophages and T cells, and promoting the production of proinflammatory cytokines, chemokines, and stress molecules via signaling pathways involving the CD74/CD44 and chemokine receptors CXCR2, CXCR4, and CXCR7 signaling. In addition, MIF can function as a stress molecule to counter-regulate the immunosuppressive effect of glucocorticoid in renal inflammation. Given the critical position of MIF in the upstream inflammatory cascade, this re view focuses on the regulatory role and molecular mechanisms of MIF in kidney diseases will be focused on.
- MIF
- inflammation
- macrophages
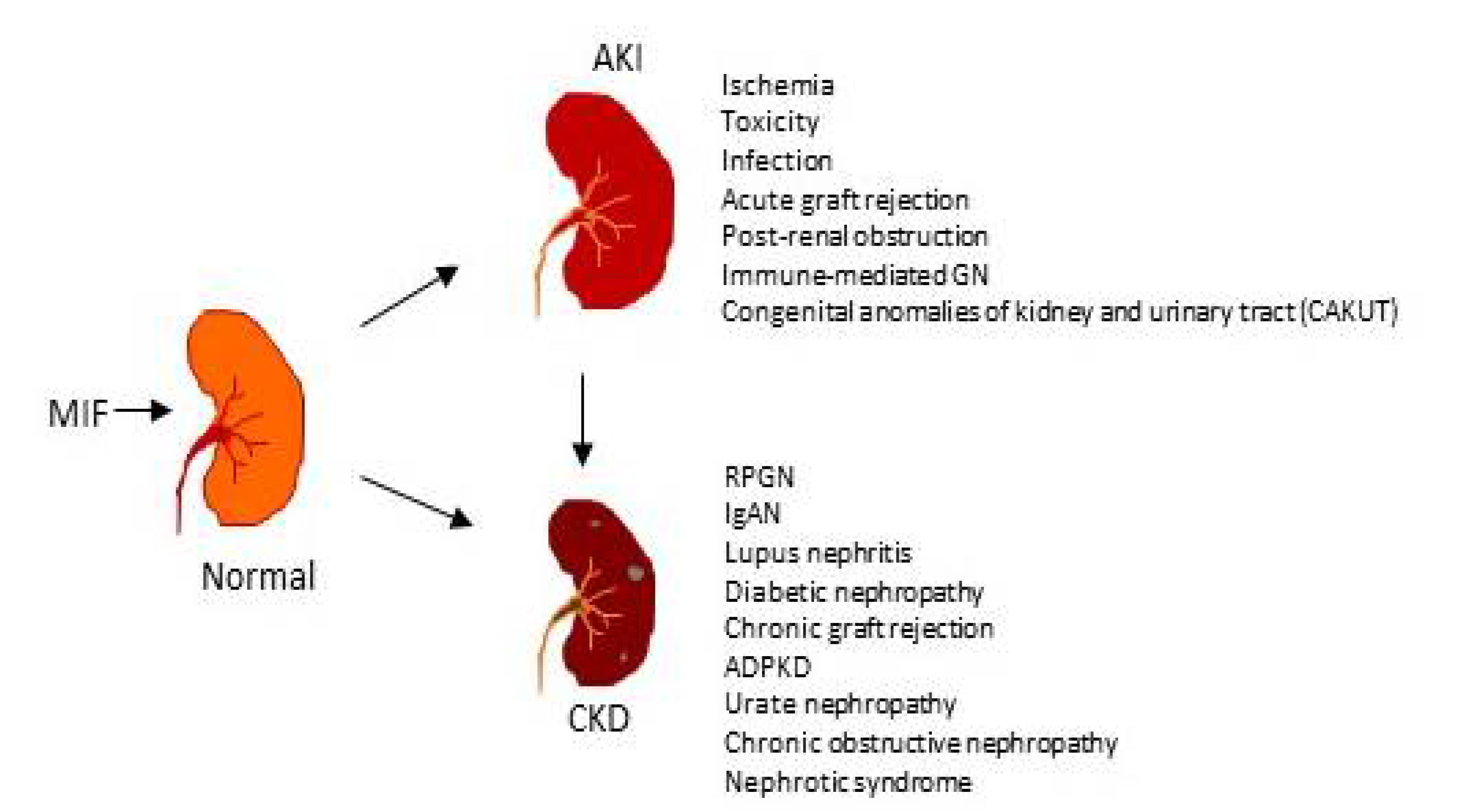
1. Role of MIF in Acute Kidney Injury
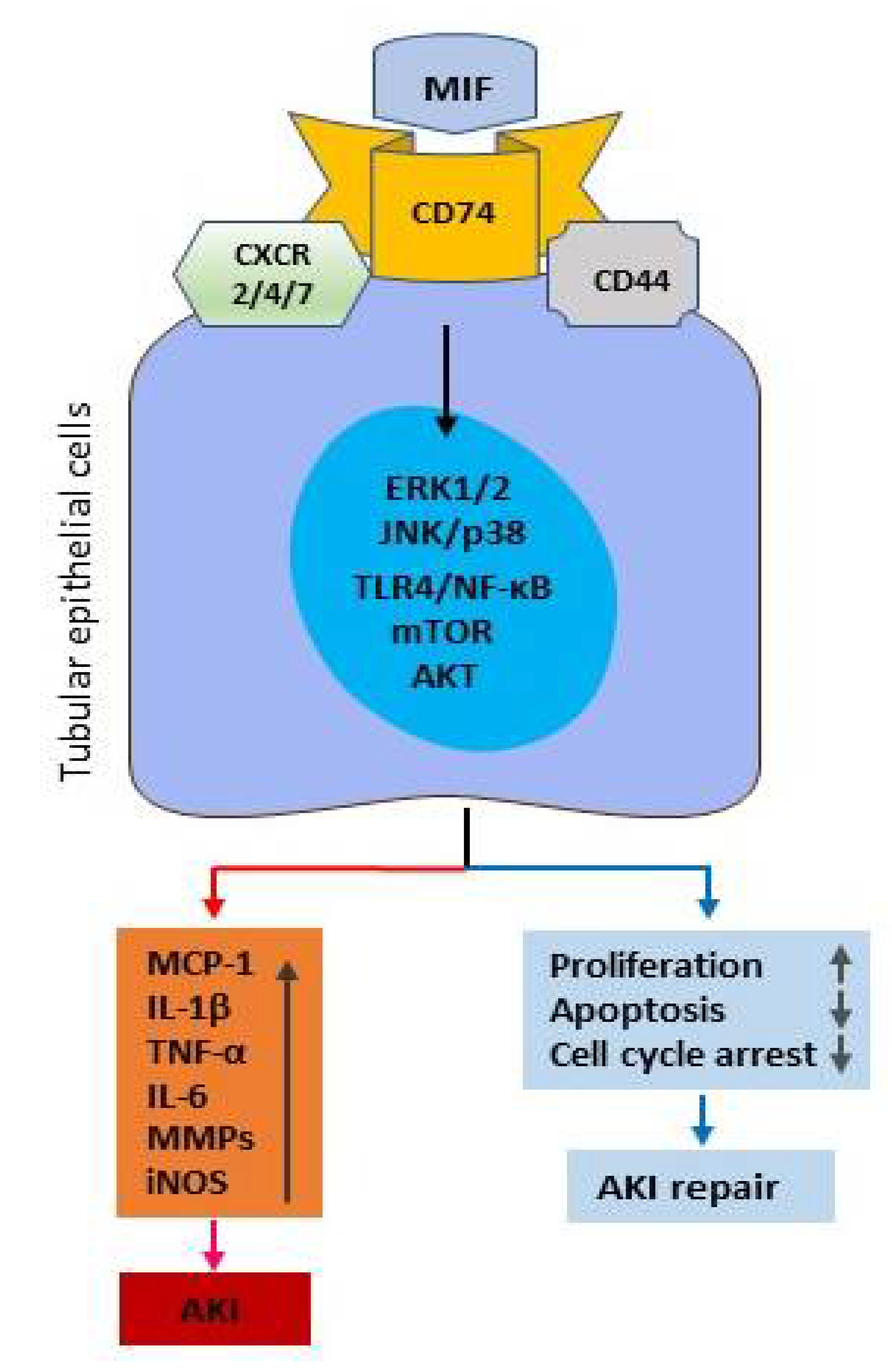
2. Mechanisms of MIF in Kidney Disease
2.1. MIF Receptors and Signal Pathways in Renal Inflammation
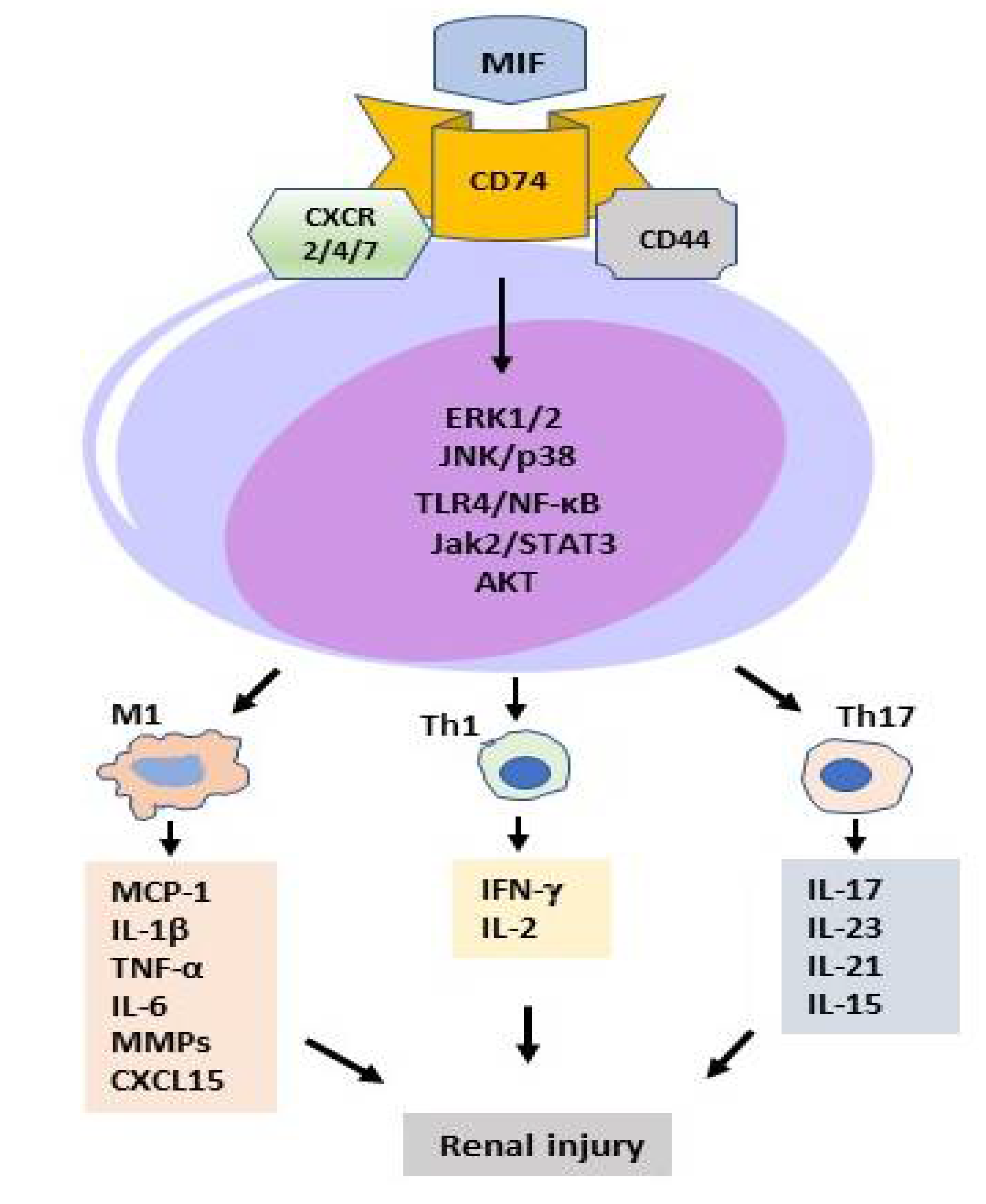
2.2. MIF in T Cell-Mediated Kidney Disease
2.3. MIF in Macrophage-Mediated Kidney Diseases
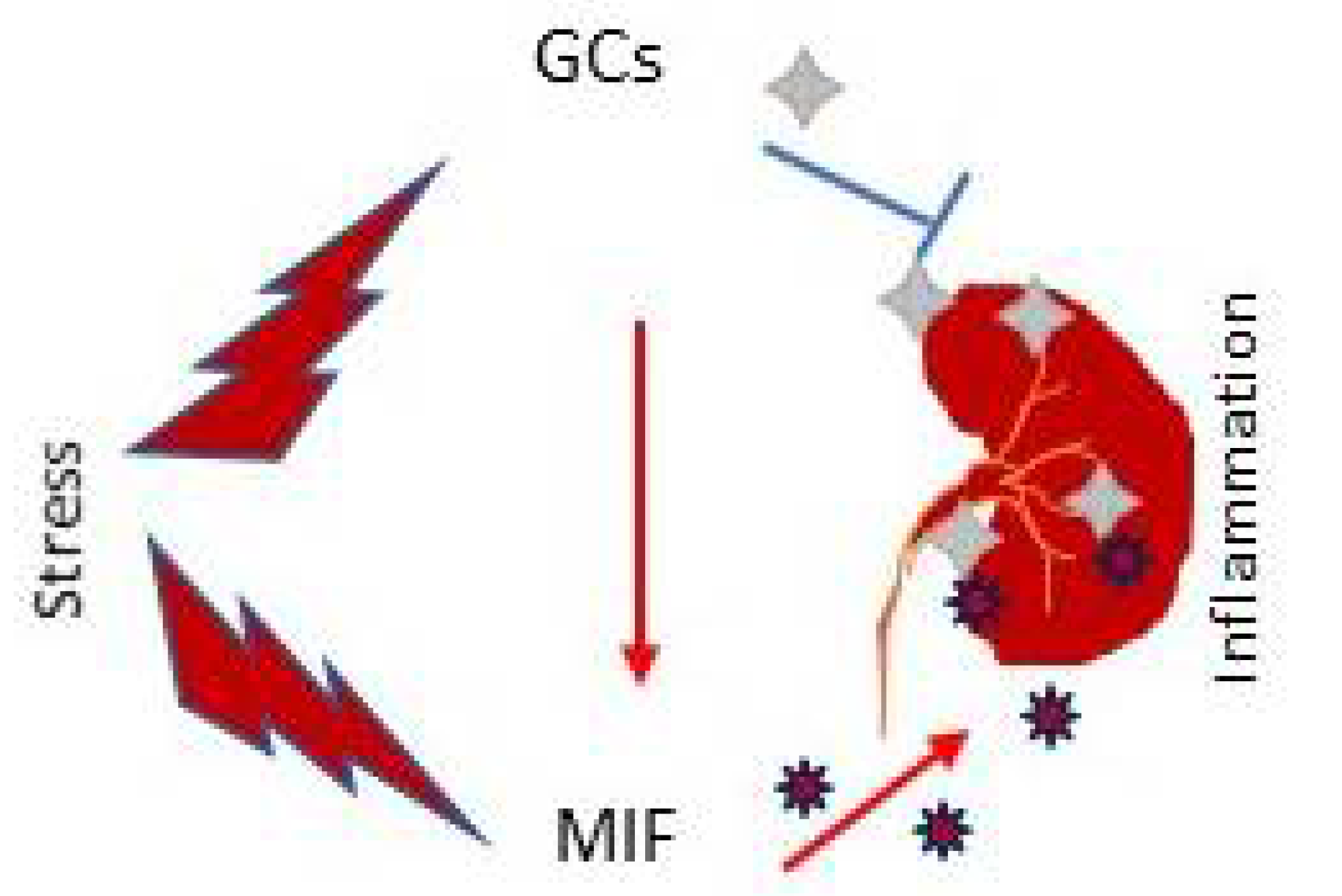
2.4. MIF as Glucocorticoid Antagonist in Renal Injury
3. MIF as a Therapeutic Target for Kidney Disease
3.1. Antibody-Based Therapy for Kidney Diseases
3.2. Treatment of Kidney Diseases with MIF Inhibitors
3.2.1. The Methyl Ester of (S, R)-3-(4-Hydroxyphenyl)-4,5-Dihydro-5-Isoxazole Acetic Acid (ISO-1)
3.2.2. Ribosomal Protein S19 (RPS19)
3.2.3. Other MIF Inhibitors
References
- Ronco, C.; Bellomo, R.; Kellum, J.A. Acute kidney injury. Lancet 2019, 394, 1949–1964.
- Hosohata, K. Role of Oxidative Stress in Drug-Induced Kidney Injury. Int. J. Mol. Sci. 2016, 17, 1826.
- Andrianova, N.V.; Zorov, D.B.; Plotnikov, E.Y. Targeting Inflammation and Oxidative Stress as a Therapy for Ischemic Kidney Injury. Biochemistry (Moscow) 2020, 85, 1591–1602.
- Guzzi, F.; Cirillo, L.; Roperto, R.M.; Romagnani, P.; Lazzeri, E. Molecular Mechanisms of the Acute Kidney Injury to Chronic Kidney Disease Transition: An Updated View. Int. J. Mol. Sci. 2019, 20, 4941.
- Baek, J.-H. The Impact of Versatile Macrophage Functions on Acute Kidney Injury and Its Outcomes. Front. Physiol. 2019, 10, 1016.
- Hong, M.-Y.; Tseng, C.-C.; Chuang, C.-C.; Chen, C.-L.; Lin, S.-H.; Lin, C.-F. Urinary Macrophage Migration Inhibitory Factor Serves as a Potential Biomarker for Acute Kidney Injury in Patients with Acute Pyelonephritis. Mediat. Inflamm. 2012, 2012, 381358.
- Payen, D.; Lukaszewicz, A.C.; Legrand, M.; Gayat, E.; Faivre, V.; Megarbane, B.; Azoulay, E.; Fieux, F.; Charron, D.; Loiseau, P.; et al. A multicentre study of acute kidney injury in severe sepsis and septic shock: Association with inflammatory phenotype and HLA genotype. PLoS ONE 2012, 7, e35838.
- Baron-Stefaniak, J.; Schiefer, J.; Miller, E.J.; Berlakovich, G.A.; Baron, D.M.; Faybik, P. Comparison of macrophage migration inhibitory factor and neutrophil gelatinase-associated lipocalin-2 to predict acute kidney injury after liver transplantation: An observational pilot study. PLoS ONE 2017, 12, e0183162.
- Pohl, J.; Hendgen-Cotta, U.B.; Stock, P.; Luedike, P.; Rassaf, T. Elevated MIF-2 levels predict mortality in critically ill patients. J. Crit. Care 2017, 40, 52–57.
- Li, T.; Zhang, J.; Long, M.; Jiang, X.; Yang, C.; Wang, F.; Su, L.; Peng, Z. Macrophage Migration Inhibitory Factor Provides A Predictive Performance of Septic Acute Kidney Injury. Shock 2022. online ahead of print.
- Stoppe, C.; Averdunk, L.; Goetzenich, A.; Soppert, J.; Marlier, A.; Kraemer, S.; Vieten, J.; Coburn, M.; Kowark, A.; Kim, B.-S.; et al. The protective role of macrophage migration inhibitory factor in acute kidney injury after cardiac surgery. Sci. Transl. Med. 2018, 10, eaan4886.
- Djudjaj, S.; Martin, I.V.; Buhl, E.M.; Nothofer, N.J.; Leng, L.; Piecychna, M.; Floege, J.; Bernhagen, J.; Bucala, R.; Boor, P. Macrophage Migration Inhibitory Factor Limits Renal Inflammation and Fibrosis by Counteracting Tubular Cell Cycle Arrest. J. Am. Soc. Nephrol. 2017, 28, 3590–3604.
- Lan, H.Y. Role of macrophage migration inhibition factor in kidney disease. Nephron. Exp. Nephrol. 2008, 109, e79–e83.
- Leng, L.; Metz, C.N.; Fang, Y.; Xu, J.; Donnelly, S.; Baugh, J.; Delohery, T.; Chen, Y.; Mitchell, R.A.; Bucala, R. MIF Signal Transduction Initiated by Binding to CD74. J. Exp. Med. 2003, 197, 1467–1476.
- Shi, X.; Leng, L.; Wang, T.; Wang, W.; Du, X.; Li, J.; McDonald, C.; Chen, Z.; Murphy, J.W.; Lolis, E.; et al. CD44 Is the Signaling Component of the Macrophage Migration Inhibitory Factor-CD74 Receptor Complex. Immunity 2006, 25, 595–606.
- Yoo, S.-A.; Leng, L.; Kim, B.-J.; Du, X.; Tilstam, P.V.; Kim, K.H.; Kong, J.-S.; Yoon, H.-J.; Liu, A.; Wang, T.; et al. MIF allele-dependent regulation of the MIF coreceptor CD44 and role in rheumatoid arthritis. Proc. Natl. Acad. Sci. USA 2016, 113, E7917–E7926.
- Bernhagen, J.; Krohn, R.; Lue, H.; Gregory, J.L.; Zernecke, A.; Koenen, R.; Dewor, M.; Georgiev, I.; Schober, A.; Leng, L.; et al. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat. Med. 2007, 13, 587–596.
- Schwartz, V.; Lue, H.; Kraemer, S.; Korbiel, J.; Krohn, R.; Ohl, K.; Bucala, R.; Weber, C.; Bernhagen, J. A functional heteromeric MIF receptor formed by CD74 and CXCR4. FEBS Lett. 2009, 583, 2749–2757.
- Alampour-Rajabi, S.; El Bounkari, O.; Rot, A.; Müller-Newen, G.; Bachelerie, F.; Gawaz, M.; Weber, C.; Schober, A.; Bernhagen, J. MIF interacts with CXCR7 to promote receptor internalization, ERK1/2 and ZAP-70 signaling, and lymphocyte chemotaxis. FASEB J. 2015, 29, 4497–4511.
- Chatterjee, M.; Borst, O.; Walker, B.; Fotinos, A.; Vogel, S.; Seizer, P.; Mack, A.; Alampour-Rajabi, S.; Rath, D.; Geisler, T.; et al. Macrophage Migration Inhibitory Factor Limits Activation-Induced Apoptosis of Platelets via CXCR7-Dependent Akt Signaling. Circ. Res. 2014, 115, 939–949.
- Sanchez-Niño, M.D.; Sanz, A.B.; Ihalmo, P.; Lassila, M.; Holthofer, H.; Mezzano, S.; Aros, C.; Groop, P.-H.; Saleem, M.A.; Mathieson, P.W.; et al. The MIF Receptor CD74 in Diabetic Podocyte Injury. J. Am. Soc. Nephrol. 2008, 20, 353–362.
- Khalilpour, J.; Roshan-Milani, S.; Gharalari, F.H.; Fard, A.A. Macrophage migration inhibitory factor antagonist (p425) ameliorates kidney histopathological and functional changes in diabetic rats. J. Bras. de Nefrol. 2019, 41, 315–322.
- Wang, Z.; Wei, M.; Wang, M.; Chen, L.; Liu, H.; Ren, Y.; Shi, K.; Jiang, H. Inhibition of Macrophage Migration Inhibitory Factor Reduces Diabetic Nephropathy in Type II Diabetes Mice. Inflammation 2014, 37, 2020–2029.
- Li, J.H.; Tang, Y.; Lv, J.; Wang, X.H.; Yang, H.; Tang, P.M.K.; Huang, X.R.; He, Z.J.; Zhou, Z.J.; Huang, Q.Y.; et al. Macrophage migration inhibitory factor promotes renal injury induced by ischemic reperfusion. J. Cell. Mol. Med. 2019, 23, 3867–3877.
- Li, J.; Tang, Y.; Tang, P.M.K.; Lv, J.; Huang, X.R.; Carlsson-Skwirut, C.; Da Costa, L.; Aspesi, A.; Frohlich, S.; Szczesniak, P.; et al. Blocking Macrophage Migration Inhibitory Factor Protects Against Cisplatin-Induced Acute Kidney Injury in Mice. Mol. Ther. 2018, 26, 2523–2532.
- Djudjaj, S.; Lue, H.; Rong, S.; Papasotiriou, M.; Klinkhammer, B.M.; Zok, S.; Klaener, O.; Braun, G.S.; Lindenmeyer, M.T.; Cohen, C.D.; et al. Macrophage Migration Inhibitory Factor Mediates Proliferative GN via CD74. J. Am. Soc. Nephrol. 2015, 27, 1650–1664.
- Shachar, I. An essential MIF-CD74 signaling axis in kidney tubular regeneration, with prospects for precision medicine and pharmacological augmentation. Am. J. Physiol. Physiol. 2017, 313, F1084–F1086.
- Chen, C.A.; Chang, J.M.; Yang, Y.L.; Chang, E.E.; Chen, H.C. Macrophage migration inhibitory factor regulates integrin-beta1 and cyclin D1 expression via ERK pathway in podocytes. Biomed. Pharm. 2020, 124, 109892.
- Safi, W.; Kraus, A.; Grampp, S.; Schödel, J.; Buchholz, B. Macrophage migration inhibitory factor is regulated by HIF-1α and cAMP and promotes renal cyst cell proliferation in a macrophage-independent manner. Klin. Wochenschr. 2020, 98, 1547–1559.
- Lan, H.Y.; Mu, W.; Yang, N.; Meinhardt, A.; Nikolic-Paterson, D.J.; Ng, Y.Y.; Bacher, M.; Atkins, R.C.; Bucala, R. De Novo renal expression of macrophage migration inhibitory factor during the development of rat crescentic glomerulonephritis. Am. J. Pathol. 1996, 149, 1119–1127.
- Lan, H.Y.; Yang, N.; Nikolic-Paterson, D.J.; Yu, X.Q.; Mu, W.; Isbel, N.M.; Metz, C.N.; Bucala, R.; Atkins, R.C. Expression of macrophage migration inhibitory factor in human glomerulonephritis. Kidney Int. 2000, 57, 499–509.
- Matsumoto, K.; Kanmatsuse, K. Increased production of macrophage migration inhibitory factor by T cells in patients with IgA nephropathy. Am. J. Nephrol. 2001, 21, 455–464.
- Lan, H.Y.; Yang, N.; Brown, F.G.; Isbel, N.M.; Nikolic-Paterson, D.J.; Mu, W.; Metz, C.N.; Bacher, M.; Atkins, R.C.; Bucala, R. Macrophage migration inhibitory factor expression in human renal allograft rejection. Transplantation 1998, 66, 1465–1471.
- Lan, H.Y.; Bacher, M.; Yang, N.; Mu, W.; Nikolic-Paterson, D.J.; Metz, C.; Meinhardt, A.; Bucala, R.; Atkins, R.C. The Pathogenic Role of Macrophage Migration Inhibitory Factor in Immunologically Induced Kidney Disease in the Rat. J. Exp. Med. 1997, 185, 1455–1466.
- De la Cruz-Mosso, U.; Garcia-Iglesias, T.; Bucala, R.; Estrada-Garcia, I.; Gonzalez-Lopez, L.; Cerpa-Cruz, S.; Parra-Rojas, I.; Gamez-Nava, J.I.; Perez-Guerrero, E.E.; Munoz-Valle, J.F. MIF promotes a differential Th1/Th2/Th17 inflammatory response in human primary cell cultures: Predominance of Th17 cytokine profile in PBMC from healthy subjects and increase of IL-6 and TNF-alpha in PBMC from active SLE patients. Cell Immunol. 2018, 324, 42–49.
- Gajic, D.; Koprivica, I.; Stojanovic, I.; Saksida, T. Defective immunosuppressive function of Treg cells in visceral adipose tissue in MIF deficient mice. Cytokine 2021, 138, 155372.
- Hoi, A.Y.; Hickey, M.J.; Hall, P.; Yamana, J.; O’Sullivan, K.M.; Santos, L.L.; James, W.G.; Kitching, A.R.; Morand, E.F. Macrophage migration inhibitory factor deficiency attenuates macrophage recruitment, glomerulonephritis, and lethality in MRL/lpr mice. J. Immunol. 2006, 177, 5687–5696.
- Yang, N.; Nikolic-Paterson, D.J.; Ng, Y.Y.; Mu, W.; Metz, C.; Bacher, M.; Meinhardt, A.; Bucala, R.; Atkins, R.C.; Lan, H.Y. Reversal of established rat crescentic glomerulonephritis by blockade of macrophage migration inhibitory factor (MIF): Potential role of MIF in regulating glucocorticoid production. Mol. Med. 1998, 4, 413–424.
- Brown, F.G.; Nikolic-Paterson, D.J.; Metz, C.; Bucala, R.; Atkins, R.C.; Lan, H.Y. Up-regulation of macrophage migration inhibitory factor in acute renal allograft rejection in the rat. Clin. Exp. Immunol. 1999, 118, 329–336.
- Lu, H.; Bai, Y.; Wu, L.; Hong, W.; Liang, Y.; Chen, B.; Bai, Y. Inhibition of Macrophage Migration Inhibitory Factor Protects against Inflammation and Matrix Deposition in Kidney Tissues after Injury. Mediat. Inflamm. 2016, 2016, 2174682.
- Lv, J.; Huang, X.R.; Klug, J.; Fröhlich, S.; Lacher, P.; Xu, A.; Meinhardt, A.; Lan, H.Y. Ribosomal protein S19 is a novel therapeutic agent in inflammatory kidney disease. Clin. Sci. 2013, 124, 627–637.
- Calandra, T.; Bucala, R. Macrophage Migration Inhibitory Factor (MIF): A Glucocorticoid Counter-Regulator within the Immune System. Crit. Rev. Immunol. 2017, 37, 359–370.
- Zaorska, K.; Zawierucha, P.; Swierczewska, M.; Ostalska-Nowicka, D.; Zachwieja, J.; Nowicki, M. Prediction of steroid resistance and steroid dependence in nephrotic syndrome children. J. Transl. Med. 2021, 19, 130.
- Cuzzoni, E.; Franca, R.; De Iudicibus, S.; Marcuzzi, A.; Lucafò, M.; Pelin, M.; Favretto, D.; Monti, E.; Morello, W.; Ghio, L.; et al. MIF plasma level as a possible tool to predict steroid responsiveness in children with idiopathic nephrotic syndrome. Eur. J. Clin. Pharmacol. 2019, 75, 1675–1683.
- Leung, J.C.; Chan, L.Y.; Tsang, A.W.; Liu, E.W.; Lam, M.F.; Tang, S.C.; Lai, K.N. Anti-macrophage migration inhibitory factor reduces transforming growth factor-beta 1 expression in experimental IgA nephropathy. Nephrol. Dial. Transplant. 2004, 19, 1976–1985.
- Jose, M.D.; David, J.R.; Atkins, R.C.; Chadban, S.J. Blockade of macrophage migration inhibitory factor does not prevent acute renal allograft rejection. Am. J. Transpl. 2003, 3, 1099–1106.
- Lubetsky, J.B.; Dios, A.; Han, J.; Aljabari, B.; Ruzsicska, B.; Mitchell, R.; Lolis, E.; Al-Abed, Y. The Tautomerase Active Site of Macrophage Migration Inhibitory Factor Is a Potential Target for Discovery of Novel Anti-inflammatory Agents. J. Biol. Chem. 2002, 277, 24976–24982.
- Leng, L.; Chen, L.; Fan, J.; Greven, D.; Arjona, A.; Du, X.; Austin, D.; Kashgarian, M.; Yin, Z.; Huang, X.R.; et al. A small-molecule macrophage migration inhibitory factor antagonist protects against glomerulonephritis in lupus-prone NZB/NZW F1 and MRL/lpr mice. J. Immunol. 2011, 186, 527–538.
- Liu, Y.; Liu, Y.; Wang, Q.; Song, Y.; Chen, S.; Cheng, B.; Zhang, Y.; Cui, Z.; Wu, Z.; Zhu, C. MIF inhibitor ISO-1 alleviates severe acute pancreatitis-associated acute kidney injury by suppressing the NLRP3 inflammasome signaling pathway. Int. Immunopharmacol. 2021, 96, 107555.
- Li, M.; Yu, J.; Zhao, L.; Mei, F.-C.; Zhou, Y.; Hong, Y.-P.; Zuo, T.; Wang, W.-X. Inhibition of macrophage migration inhibitory factor attenuates inflammation and fetal kidney injury in a rat model of acute pancreatitis in pregnancy. Int. Immunopharmacol. 2019, 68, 106–114.
- Li, T.; Sun, H.; Li, Y.; Su, L.; Jiang, J.; Liu, Y.; Jiang, N.; Huang, R.; Zhang, J.; Peng, Z. Downregulation of macrophage migration inhibitory factor attenuates NLRP3 inflammasome mediated pyroptosis in sepsis-induced AKI. Cell Death Discov. 2022, 8, 61.
- Sanchez-Zamora, Y.; Terrazas, L.I.; Vilches-Flores, A.; Leal, E.; Juárez, I.; Whitacre, C.; Kithcart, A.; Pruitt, J.; Sielecki, T.; Satoskar, A.R.; et al. Macrophage migration inhibitory factor is a therapeutic target in treatment of non-insulin-dependent diabetes mellitus. FASEB J. 2010, 24, 2583–2590.
- Zheng, L.; Gao, J.; Jin, K.; Chen, Z.; Yu, W.; Zhu, K.; Huang, W.; Liu, F.; Mei, L.; Lou, C.; et al. Macrophage migration inhibitory factor (MIF) inhibitor 4-IPP suppresses osteoclast formation and promotes osteoblast differentiation through the inhibition of the NF-kappaB signaling pathway. FASEB J. 2019, 33, 7667–7683.
- Zheng, L.; Feng, Z.; Tao, S.; Gao, J.; Lin, Y.; Wei, X.; Zheng, B.; Huang, B.; Zheng, Z.; Zhang, X.; et al. Destabilization of macrophage migration inhibitory factor by 4-IPP reduces NF-kappaB/P-TEFb complex-mediated c-Myb transcription to suppress osteosarcoma tumourigenesis. Clin. Transl. Med. 2022, 12, e652.
- Jin, K.; Zheng, L.; Ye, L.; Xie, Z.; Gao, J.; Lou, C.; Pan, W.; Pan, B.; Liu, S.; Chen, Z.; et al. Chicago sky blue 6B (CSB6B), an allosteric inhibitor of macrophage migration inhibitory factor (MIF), suppresses osteoclastogenesis and promotes osteogenesis through the inhibition of the NF-kappaB signaling pathway. Biochem. Pharmacol. 2021, 192, 114734.