Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Siyuan Kong and Version 2 by Rita Xu.
Nucleosome-Omics is one of the subdisciplines of Omics, which studies nucleosome-level phenomenon on chromatin and genome 3D landscape, including the interaction and combination among histones, trancriptional factors and DNA, by combining nucleosome resolution omics technologies with high-throughput sequencing techniques.
- Nucleosome-Omics
- epigenetics
- nucleosomes
1. Introduction of Nucleosome-Omics
The nuclear genetic code of eukaryotes (such as animals and plants) depends on chromatin, which is a highly organized complex of DNA and proteins and is a principal component of the cell nucleus [1]. What is amazing is that chromatin almost carries most of the genetic information about organic development, growth, and aging. With conventional electron microscopy measurements, it was found that chromatin containing roughly 2 m DNA is condensed and folded in the nucleus, which is only about 10 μm in diameter [2]. The diameter of a classical chromatin fiber is just only ~30 nm [3]. Scientists have reached a consensus that nucleosomes could be thought of as the basic building blocks of chromatin [4]. In addition, the “beads on a string” conformation vividly depicts the state of nucleosomes in chromatin [5]. During the different biological processes, such as cell division, cell proliferation, and differentiation, the “beads on a string” nucleosomes can flexibly compress and loosen with DNA duplication and transcriptional regulation. Nucleosomes are basal structural units, which consist of histone proteins and organizers. They are assembled into compact structures called chromatin in interphase, or into enormous, high-order structures called chromosomes in mitosis [1]. Scientists have used classical physics tools and methods, electron microscopy, X-ray scattering and crystallography, etc., and found or proposed some models [3]. For example, from the 1970s–2010s, there are the one-start solenoid model [6], a two-start helical ribbon model [7], a two-start crossed-linker model [8], a polymer melt model [9], and a two-start zig-zag model with tetranucleosomal unit [10]. This makes the nucleosome folding structure and pattern more and more clear [3]. Therefore, in the early days of nucleosome research, especially twenty years ago, these studies mainly focused on ultrastructural observation and structural model analysis [3]. However, the effects of chromatin conformation and nucleosome dynamics on transcriptional regulation have been poorly studied and unclear. Recently, with the development of next-generation sequencing (NGS) technology, various nucleosome-resolution or even base-pair resolution-level genomics sequencing technologies have been developed in succession. For instance, based on nucleosome-level genome fragment digestion by micrococcal nuclease (MNase), MNase-seq and derivative technology [11], MNase-Exo-seq [12], MNase-ChIP-seq [13], MACC-seq [14], etc. was developed. With these technologies or integrating with other omics technologies (ChIP-seq, RNA-seq, or others), accurate nucleosome position, the distribution of specific genomic regions, and their impacts on transcriptional regulation and gene expression were revealed [15]. Fortunately, it is worth pointing out that the development of three-dimensional (3D) genomics technologies makes it possible to analyze the three-dimensional conformations of chromatin. Hi-C and ChIA-PET are the representative technologies that burst the field of epigenetic research [16]. By combining Hi-C with MNase genome fragmentation, Micro-C and its derivative Micro-C XL can be improved [17]. These technologies help unmask ultra-fine-scale chromatin ultrastructure at the 100–1000 base-pair resolution [18]. Some special structures, dots, and stripes, which are not easily found in the Hi-C, are well represented and reconstructed [19].
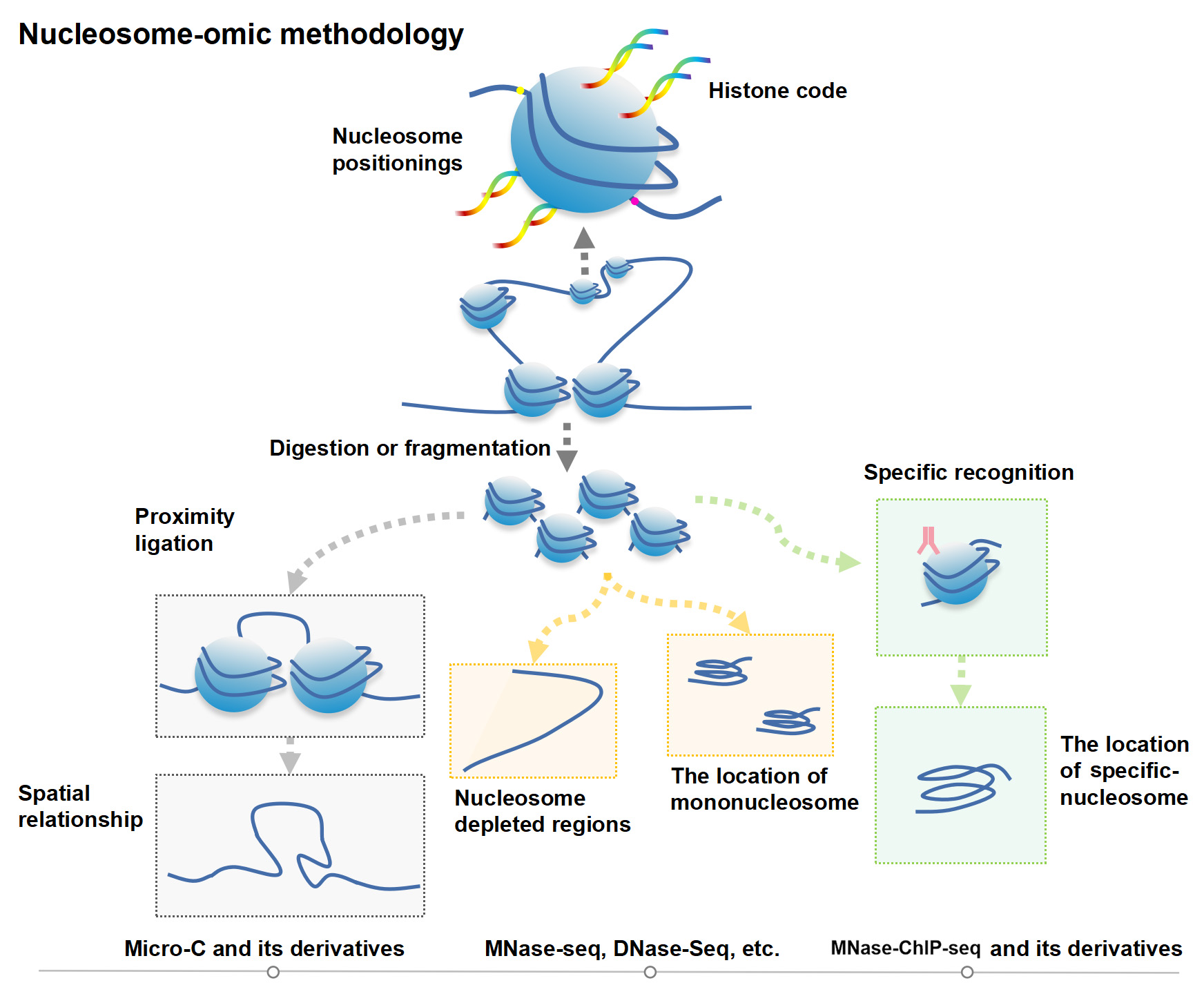
Figure 1.
Nucleosome-moic methodology.
2. Extended information
2.1. Scientific Discovery, Development and Related Research of Nucleosomes
Chromatin is the carrier of genetic and epigenetic information about organisms. Under an optical microscope, at the 1400 nM length interval, rwesearchers could observe some regular-shaped and compact chromosome packages called metaphase chromosomes, and sometimes, loose and seemingly irregular chromatin in other interphase cells [20]. The chromosome packages the genome via a hierarchical series of folding steps ranging from 700 nM, 300 nM, to 30 nM (Figure 2a) [6]. In addition, the chromatin can directly extend 30 nM chromatin fibrils (Figure 2a) [20]. Thirty nM chromatin fiber’s substructure and its higher-order structure are hot spots in the current spatial chromatin conformation epigenetics research [10]. Physical and biochemical approaches complement each other to study the chromatin spatial conformation [3]. By using microscopic imaging techniques, ChromEMT, the nucleus, chromatins, nucleosomes, and chromatin loops are visualized [20].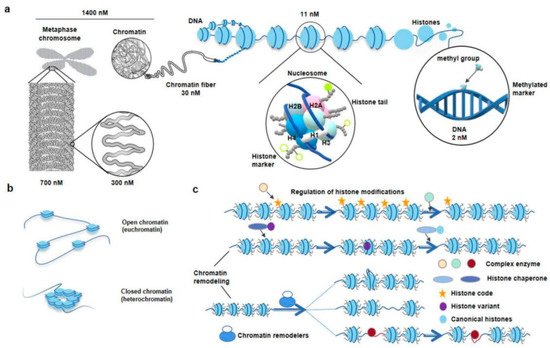
Figure 2. The diagram of hierarchical structures from chromatin to nucleosome in eukaryotes. (a), Zoom in chromosome or chromatin to DNA. The DNA double helix wraps around histone octamers to form nucleosomes, the basic structural units of chromatin. The classic histone octamer is made up of four types of histone (H2A/H2B/H3/H4) and has eight histone tails. Covalent modification markers on histone tails play an important role in regulating the chromatin structure and function. DNA methylation markers are also epigenetic codes. (b), Different states of chromatin. The regions of chromatin, called euchromatin, exist in an extended and loosely packed state, which is conducive to transcription. Conversely, densely packed heterochromatin is silent. (c), Regulation of histone modifications and chromatin remodeling. Recognition, reading, and removal of histone modifications depend on the Writer, Reader, and Eraser. The complex enzymes responsible for the alteration of histone modifications often have more than one ability. When the reading modules of these complexes bind to the corresponding sites, their writing or erasure modules are activated and work at nearby sites. ATP-driven chromatin remodelers, which are complex of multiple subunits, catalyze chromatin remodeling. Meanwhile, the nucleosome structure usually changes in four ways: replacement, dissociation, removing, and slide. Replacement indicates chromatin remodelers, which catalyze replacement between canonical histone and histone variants in nucleosomes. Dissociation indicates that the double helix DNA wrapped around the nucleosome loosens. Removing indicates the disintegration of nucleosomes. Slide indicates chromatin remodelers can allow nucleosomes to slide along DNA without unwinding the DNA double strand [21][32]. Courtesy: National Human Genome Research Institute (https://www.genome.gov/genetics-glossary/Chromatin, accessed on 19 May 2022; https://www.genome.gov/genetics-glossary/Nucleosome, accessed on 19 May 2022).