Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 2 by Jessie Wu and Version 1 by Eleftheria Galatou.
Nonalcoholic steatohepatitis (NASH) is the most severe manifestation of nonalcoholic fatty liver disease (NAFLD), a common complication of type 2 diabetes, and may lead to cirrhosis and hepatocellular carcinoma. Oxidative stress and liver cell damage are the major triggers of the severe hepatic inflammation that characterizes NASH, which is highly correlated with atherosclerosis and coronary artery disease.
- nonalcoholic steatohepatitis (NASH)
- nonalcoholic fatty liver disease (NAFLD)
- atherosclerosis
1. Diagnosis of Nonalcoholic Fatty Liver Disease/Nonalcoholic Steatohepatitis
NAFLD comprises a wide spectrum of liver diseases ranging from simple fat accumulation to more advanced stages, including NASH, cirrhosis, and cancer. The diagnosis of NAFLD requires the identification of hepatic steatosis (liver fat > 5%) in the absence of other causes of liver fat accumulation (e.g., alcohol consumption or coexisting causes of chronic liver diseases) [9,14][1][2]. NAFLD is often suspected in clinical practice when an individual presents features of MetS, such us abdominal obesity, hypertension, increased levels of triglycerides, low levels of HDL, and increased levels of fasting blood glucose. In the same context, individuals with persistently abnormal liver enzyme levels should be screened for NAFLD/NASH [9][1]. Several studies have shown that mild-to-moderate elevations in serum liver enzymes or increased liver volume [15][3] are associated with a higher risk of all-cause mortality [16,17,18][4][5][6]. There are many available imaging techniques, including ultrasound, image-guided biopsy, computed tomography (CT), and magnetic resonance imaging (MRI). Multiparametric MRI combines two or more quantitative techniques, such as T1, T2, and the proton density fat fraction (PDFF), to assess hepatic inflammation and fibrosis with a high level of accuracy. MRI-PDFF was more accurate at detecting changes in liver fat than liver biopsy and has been validated in multiple studies [19,20,21][7][8][9]. Ultrasound is widely accepted as a first-line diagnostic tool since it is a non-invasive, low cost, and radiation-free technique with a satisfactory sensitivity for moderate and severe steatosis identification [22][10]. To date, the gold standard to diagnose patients with NASH is still considered to be liver biopsy, demonstrating the typical fibrosis pattern, which cannot be seen via imaging methods. Currently, although there is no readily available, reliable, and non-invasive method to identify the progression of steatosis to NASH and fibrosis, altered levels of lipoprotein (a) (Lp(a)) [23][11] and several biomarkers of inflammation (such as ferritin and high-sensitivity C reactive protein (CRP)) and apoptosis (cytokeratine 18 (CK-18)) [24][12] have been associated with the diagnosis of NASH in NAFLD patients. Moreover, the NAFLD Activity Score (NAS) is used in clinical trials for evaluating the changes in histological features caused by therapeutic interventions. This system was developed and validated by the Pathology Committee of the NASH Clinical Research Network (NASH CRN) and is based on the semi-quantitative evaluation of histological features, specifically steatosis (0–3), lobular inflammation (0–2), and hepatocellular ballooning (0–2). NAS is calculated by adding these values, with a sum of ≥5 indicating NASH and scores < 3 considered as “not NASH” [25][13]. However, the threshold value of 5 in NAS is not always in accordance with a NASH diagnosis that is based on the analysis of a liver biopsy for the existence of certain lesions with specific patterns [26][14].
2. Pathophysiological Mechanisms of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis
NAFLD is a metabolic disorder, and its pathogenesis is a multifactorial process that involves a complex interaction between metabolic, clinical, environmental, and genetic factors [38,39][15][16].
2.1. Nutrition and Gut Microbiota
Components of nutrition and caloric intake play a key role in NAFLD development and progression. A nutritional high-fat and hypercaloric pattern rich in saturated fat and omega-6 (n-6) polyunsaturated fatty acids (PUFAs), carbohydrates, and low amounts of omega-3 (n-3) PUFAs and fibers have all been associated with NAFLD [40][17]. Data from preclinical and clinical studies demonstrate that fructose intake in hypercaloric diets is associated with an increased intrahepatic content of triglycerides, de novo lipogenesis, hepatic steatosis, obesity, and insulin resistance [41,42][18][19]. Glucose intake exerts similar detrimental effects on liver health by increasing hepatic lipid accumulation in healthy men [43][20]. This energy imbalance leads to higher post-prandial blood glucose levels and, thus, a higher insulin secretion rate. Insulin further stimulates de novo lipogenesis, leading to hepatic inflammation and subsequent NASH development. It has been demonstrated that a high-fat diet induces hepatic tumor necrosis factor-a (TNF-a) and interleukin (IL)-6 expression, whereas their inhibition prevents hepatic steatosis and NAFLD progression.
The body of evidence supporting an association between gut microbiota and NAFLD pathogenesis and progression is increasing. Basic players for complex dietary carbohydrate degradation are gut bacteria, leading to the production of short-chain fatty acid (SCFA) metabolites, including acetate, propionate, and butyrate. Although these metabolites improve glucose and lipid metabolism and maintain intestinal homeostasis, the increased production may contribute to obesity and liver steatosis, enhancing nutrient absorption [44][21]. Moreover, bile acids, which are synthesized by cholesterol within the liver, metabolized in the small intestine, and reabsorbed back to the liver through the portal vein, are significant regulators of lipid and carbohydrate metabolism and, subsequently, energy homeostasis. In the liver, they act as signaling molecules through their binding and activation of several nuclear hormone receptors, including farnesoid X receptors (FXRs) and G-protein-coupled receptor 5 (TGR5) [45][22]. Many studies have assessed the interplay between bile acids and microbiota, demonstrating that bile acids regulate and control the microbiome, while gut bacteria contribute to several biotransformations of bile acids and affect their composition, modulating hepatic steatosis [46,47,48,49][23][24][25][26].
It is well-documented that patients with biopsy-proven NAFLD/NASH exhibit a different microbiota signature and increased gut permeability due to a disruption of gut epithelial tight junctions. The leaky intestine leads to a translocation of bacteria and bacteria-produced endotoxins and alcohol through the portal circulation to the liver, contributing to ROS generation, hepatic inflammation through the toll-like receptor (TLR)-4-mediated pathway, and possibly fibrogenesis [49,50,51][26][27][28].
2.2. Adipose Tissue Dysfunction—The Role of Adipokines
Adipose tissue is an essential and highly active metabolic and endocrine organ that stores triacylglycerol as an energy source and releases adipokines and cytokines that are suspected to play a key role in NAFLD development and the progression to NASH. Visceral adiposity leads to excessive lipid accumulation and is highly correlated with insulin resistance due to an imbalance in pro-inflammatory and anti-inflammatory cytokine release. Leptin, an adipokine that plays a crucial role in the regulation of body weight and fat content, primarily acts centrally to reduce food intake, increase energy expenditure, and prevent lipid accumulation in organs other than the adipose tissue. However, excessive levels of leptin may result in hepatic inflammation and fibrosis [52][29]. Adiponectin, an abundant adipokine with anti-inflammatory and antifibrotic properties that acts on Kupffer cells and hepatic stellate cells (HSCs), increases hepatic insulin sensitivity by suppressing gluconeogenesis and lipogenesis and reduces body fat [50][27]. The anti-inflammatory effects of adiponectin are achieved by: (1) the suppression of transcription factor NF-κB, (2) the inhibition of pro-inflammatory cytokine release, and (3) the stimulation of anti-inflammatory cytokine secretion [51][28]. Patients with NAFLD and NASH exhibit elevated leptin levels and decreased adiponectin levels, which are associated with the severity of NAFLD patients, probably reflecting the increasing insulin resistance [8,52][29][30]. However, in later stages of NASH progression to cirrhosis, adiponectin levels are increased, possibly due to an impaired clearance of adiponectin and an excessive release of pro-inflammatory cytokines.
2.3. Insulin Resistance and Hepatic Fat Accumulation
Two pivotal characteristics of NAFLD pathophysiology are insulin resistance and hepatic steatosis. Insulin plays a crucial role in the regulation of glucose and lipid metabolism in several metabolic tissues, including adipose tissue and the liver. In hepatocytes, insulin regulates glucose uptake, promotes glycogenesis, and activates key regulators of de novo lipogenesis (DNL) while it simultaneously decreases gluconeogenesis, promoting glycogen storage [53][31]. On the other hand, in adipocytes, insulin has three main actions: (1) to promote the esterification of fatty acids, (2) to promote the storage of esterified fatty acids, including triglycerides (TGs), in lipid droplets, and (3) to inhibit lipolysis via hormone-sensitive lipase inactivation [54][32].
In an obese state and in NAFLD patients, systemic insulin resistance results in increased lipolysis and, thus, excess free fatty acids (FFAs), and inflammatory cytokines from peripheral adipose tissue can enter the liver through the portal circulation. The accumulation of FFAs and lipid metabolites in hepatocytes may induce a disruption of the insulin-signaling pathway and, subsequently, hepatic insulin resistance. Moreover, hepatic insulin resistance contributes to hyperglycemia, hyperinsulinemia, and increased lipid accumulation through DNL stimulation and mitochondrial fatty acid β-oxidation (FAO) inhibition, thus aggravating hepatic steatosis [55,56][33][34].
2.4. Progression of NAFLD to NASH
2.4.1. Lipotoxicity and Oxidative Stress
Several studies have highlighted that lipotoxicity leads to hepatocyte injury and the progression of NASH. The imbalance between lipid acquisition (increased uptake of circulating FFAs and DNL) and lipid exportation (downregulated FAO and export of lipids in very low density lipoproteins (VLDL)) further promotes lipid accumulation in the liver and the progression of hepatic steatosis [56,57,58][34][35][36]. In addition to FFAs, other types of lipids and their derivatives, including free cholesterol and ceramides, are involved in the development of liver lipotoxicity in NAFLD and NASH patients [59,60][37][38]. Lipotoxicity-induced hepatic injury leads to hepatocyte ballooning degeneration (liver cell swelling), fibrosis, and glomerular inflammation, which are considered to be the key histological features for NASH diagnosis [61,62][39][40].
In NAFLD patients, mitochondrial dysfunction plays a pivotal role during the transition from simple steatosis to NASH [63][41]. The energy homeostasis in hepatic cells is regulated by mitochondrial FAO, electron transfer, the production of ATP, and reactive oxygen species (ROS) [64][42]. Mitochondrial dysfunction contributes to an imbalance between prooxidant and antioxidant mechanisms, thus leading to lipid accumulation and excess ROS generation. The latter leads to the activation of inflammatory mediators and signaling pathways exacerbating inflammation, ROS generation, and oxidative DNA damage in NASH patients [65,66][43][44]. The overload of FFAs leads to an increase in the permeability of the inner mitochondrial membrane, 31–40% lower maximal respiration associated with mitochondrial uncoupling, electron leakage, augmented hepatic oxidative stress, and oxidative DNA damage [4,65][43][45].
2.4.2. Hepatic Inflammation and Fibrosis
The mechanistic concepts of inflammation in NAFLD/NASH have recently been reviewed and are associated with the exacerbated production of inflammatory factors from extrahepatic tissues (adipose tissue and gut) and in the liver by injured hepatocytes and the activation of resident hepatic macrophages, Kupffer cells [67][46]. Excess levels of circulating and hepatic FFA accumulation, altered gut microbiome, gut permeability alterations, the release of endotoxin, and adipose tissue dysfunction lead to hepatocyte injury and apoptosis, ROS generation, and inflammatory response, representing the initial steps of progression to NASH. Immunogenic stimuli, including damage-associated molecular patterns (DAMPs) released by injured hepatocytes and pathogen-associated molecular patterns (PAMPs), are recognized by the innate immune system through pattern recognition receptors (PRRs), such as toll-like receptors (TLRs) and NOD-like receptors (NLRs), and play a key role in NASH [68][47]. Being widely expressed in hepatic cells, activated TLRs, particularly TLR-4, recruit Kupffer cells, which release pro-inflammatory cytokines, including TNF-a, IL-1, IL-6, fibrogenic factors such as TGF-β, and the further activation of pro-inflammatory transcription factors (NF-κB) [69][48]. Cytokine signaling promotes the recruitment of immune effector cells, including neutrophils, dendritic cells, natural killer (NK) cells, and cytotoxic T cells, with subsequent hepatocyte injury via oxidative-stress-mediated mechanisms, indicating that adaptive immunity plays an important role in the progression of this disease [70][49]. In NASH patients, NOD-like receptor protein 3 (NLRP3) inflammasome is upregulated in injured hepatocytes, Kupffer cells, and liver sinusoidal endothelial cells and can be activated by DAMPs and PAMPs [71,72][50][51].
Chronic inflammation is associated with fibrosis, which can further progress to bridging fibrosis and cirrhosis. In response to liver injury, activated Kupffer cells, infiltrating monocytes, activated and aggregated platelets, and damaged hepatocytes release platelet-derived growth factor (PDGF) and TGF-β1, leading to HSC activation. Upon their activation, HSCs express NLRP3 inflammasome and transdifferentiate into fibroblasts or myofibroblast-like cells with proliferative, inflammatory, and migratory properties [73][52]. Upon HSC proliferation, components of the extracellular matrix are produced, including collagen type I and type III as well as tissue inhibitor of metalloproteinases 1 (TIMP-1), all contributing to fibrogenesis [74,75][53][54].
3. Association between NASH and Atherosclerosis: Inflammation and Oxidative Stress as Key Players
A large body of epidemiological and clinical evidence demonstrates that NAFLD is not only associated with liver morbidity but also with CVD development, arrhythmias, left ventricular dysfunction, and heart failure [76,77,78][55][56][57]. NAFLD and CVD are both manifestations of end-organ damage of MetS and share several common environmental and genetic factors [79,80,81][58][59][60] It has been demonstrated that NAFLD progression to NASH is associated with more severe atherosclerosis and is even considered an independent risk factor for coronary artery disease (CAD) [8,82][30][61]. Likewise, atherosclerosis accompanied by NAFLD/NASH has more adverse metabolic burdens than atherosclerosis alone. In this aspect, several studies provide evidence of a strong association between NASH and: (1) carotid atherosclerosis [83,84,85,86][62][63][64][65] and (2) subclinical manifestations of atherosclerosis in patients with or without T2DM, including increased intima–media thickness, endothelial dysfunction, arterial stiffness, impaired left ventricular function, reduced flow-mediated vasodilation, and coronary calcification [86,87,88][65][66][67]. These associations are independent of traditional cardiovascular risk factors and MetS characteristics across a wide range of patient populations [89][68]. Atherosclerosis is a progressive multifactorial disease characterized by the thickening of the arteries and endothelial dysfunction and is a main cause of myocardial infarction and stroke [90,91][69][70] Proinflammatory activation of endothelial cells (ECs) initiate the penetration of monocytes into the intima media, where they predominantly mature to pro-inflammatory macrophages (the M1 phenotype) that actively take up modified low-density lipoproteins (oxLDL) via scavenger receptors (e.g., scavenger receptor A1 (SR-A1), lectin-like oxLDL receptor-1 (LOX-1), and CD36) and release a variety of inflammatory cytokines and chemokines that are essential for the propagation of inflammation [92][71]. The excessive influx of modified LDLs and the accumulation of cholesterol esters in intimal macrophages due to the increased vascular permeability of the endothelial barrier lead to the generation of foam cells, which play a key role at all stages of atherosclerotic lesion development, from initial vascular lesions to advanced plaques [91][70]. Another main imbalance observed in atherosclerosis is the upregulation of acetyl-CoA acetyltransferase (ACAT1), the enzyme responsible for cholesterol esterification, and the downregulation of neutral cholesterol ester hydrolase (NCEH), the enzyme responsible for the hydrolysis of cholesterol esters to free cholesterol, resulting in the accumulation of cholesterol esters and the further transformation of macrophages to foam cells. Yellow foam cells aggregate on the arterial walls and cause the development of fatty streaks, which form a fibrous atherosclerotic plaque cap [93,94][72][73]. At advanced stages of the disease, growth factor released by macrophages in the plaque causes the proliferation of smooth muscle cells and the plaque becomes fibrotic [95][74]. Activated macrophages and T lymphocytes of the fibrous atherosclerotic plaque cap stimulate the production of proteolytic metalloproteases, leading to the degradation of the extracellular matrix by phagocytosis and to a decrease in the stability of the fibrous cap. Plaque rupture leads to a coagulation process, blood clot formation, thrombus formation, and a blockade of the arteries [96][75]. These atherogenic processes are triggered by well-identified risk factors, such as hypertension, hyperlipidemia, and diabetes mellitus. NAFLD can contribute to and aggravate atherosclerosis development, but the precise mechanisms remain unclear. The supposed mechanisms for accelerating atherosclerotic disease in patients with NASH are very complex and include, among others, chronic inflammation, lipid accumulation, and oxidative stress (Figure 1). The following sections display possible linkages between these conditions at the molecular level.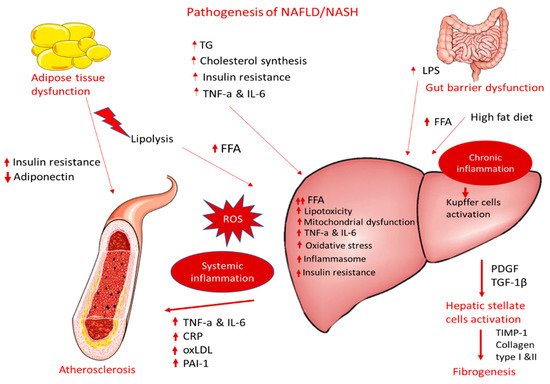
Figure 1.
Pathogenesis of NAFLD/NASH and association with atherosclerosis.
3.1. Dyslipidemia and Lipotoxicity
As mentioned before, NAFLD/NASH is characterized by hepatic fat accumulation, which results from an imbalance between lipid acquisition and lipid disposal that is mediated by increased hepatic DNL and the uptake of circulating FFAs, a downregulation of compensatory FAO, and an altered export of lipids in VLDL. This imbalance is considered to be the initiating mechanism of atherosclerosis as well [97][76]. Moreover, patients with NAFLD exert typical atherogenic dyslipidemia features, including higher serum TG and oxLDL levels and lower serum HDL. Moreover, high serum oxLDL levels are more dominant in patients with atherosclerosis due to their localization in macrophage-derived foam cells. It has been reported that TNF-a is implicated in the decrease in HDL levels, indicating a link between inflammation and the development and progression of insulin-resistant conditions, including NASH and atherosclerosis [98,99][77][78].3.2. Adipose Tissue Dysfunction
Adipose tissue increases CVD risk by inducing many obesity-associated complications, such as dyslipidemia, high blood pressure, insulin resistance, and T2DM [100][79]. Visceral adiposity causes an increase in the hepatic accumulation of FFAs, accompanied by decreased FFA oxidation and altered glucose metabolism, contributing to hepatic insulin resistance. Visceral obesity is associated with proinflammatory cytokine production, i.e., TNF-α, IL-6, monocyte chemoattractant protein-1 (MCP-1), CRP, adipokines, and macrophage infiltration, resulting in local and systemic inflammation. The latter is associated with the consequent hepatic production of pro-atherogenic molecules, such as plasminogen activator inhibitor-1 (PAI-1) and fibrinogen, thereby causing endothelial dysfunction and increasing the risk of atherothrombosis. A consistent and ever-growing line of research has revealed a link between intestinal dysbiosis, inflamed adipose tissue, and NAFLD with atherosclerosis and other cardiac complications. The progression of NAFLD to NASH leads to the production of proinflammatory cytokines, atherogenic lipoproteins, and vasoactive and thrombogenic factors and enhanced oxidative stress, resulting in an increased risk of atherosclerosis and myocardial infarction.3.3. Endothelial Dysfunction and Inflammation
The endothelial dysfunction and abnormal vasoreactivity observed in NASH patients lead to chronic inflammation, increased vasoconstriction, and increased prothrombotic factor production, thus elevating the risk of atherosclerosis and several other cardiovascular implications [101][80]. Inflammation plays a central role in both NASH and atherosclerosis, involving the local presence or resident macrophages. Macrophages accumulate oxidized lipoproteins through the SRs and lead to the generation of foam cells and the release of cytokines during atherogenesis. Similarly, in NASH the resident hepatic macrophages, Kupffer cells, take up modified lipoproteins expressing SR CD36 and, thus, further contribute to atherosclerotic lesions [93,99][72][78]. Specifically, immunogenic stimuli, including the accumulation of modified triglyceride-rich lipoproteins, serve as DAMPs and activate TLRs recruiting macrophages and resident Kupffer cells. The activated NLRP3 inflammasome leads to the release of pro-inflammatory cytokines, including TNF-a, IL-1, and IL-6; fibrogenic factors, such as TGF-β; and the further activation of pro-inflammatory transcription factors (NF-κB) [69][48], providing an important link between NASH, liver fibrosis, and the development of vascular damage and atherosclerosis [102][81].4. Explaining the Effectiveness of Incretin-Based Drugs on NASH and Atherosclerosis
4.1. Potential Mechanisms of GLP-1 RAs’ Effectiveness in NASH
The only GLP-1 RAs that have been proven to resolve NASH based on histological data are liraglutide and semaglutide. These GLP-1 RAs, apart from their glucose-lowering effect, have demonstrated an ability to significantly reduce body weight, with semaglutide having a more pronounced effect. This effect is mainly attributed to a modulation of appetite and a feeling of satiety as well as reduced caloric intake through actions in the central nervous system combined with a reduction in glucosuria due to enhanced glycemic control (Figure 2) [145][82]. Most of the weight that is being lost during treatment with GLP-1 RAs is fat mass, particularly visceral fat, due to their effect on adipose tissue [146][83]. However, although body weight reduction is a key parameter in NASH resolution, it cannot solely explain the improved liver function observed in patients treated with GLP-1 RAs. Specifically, in a study performed by Shiomi et al. (2020) in Japanese patients with T2DM and NAFLD receiving liraglutide for 24 weeks the improvement in liver function or fibrosis (assessed through aspartate aminotransferase, alanine aminotransferase, and fibrosis-4 indices) was found to be independent of the body mass index [147][84]. Several other mechanisms are involved, as demonstrated in Figure 2.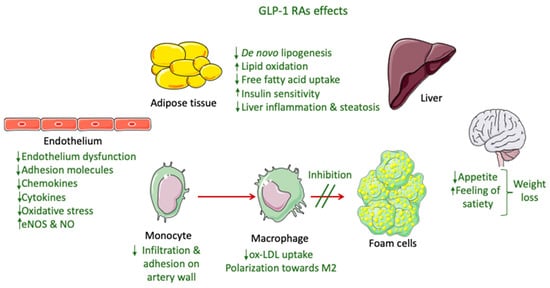
Figure 2.
GLP-1 RA effects involved in NASH and atherosclerosis improvement.
4.2. Potential Mechanisms of GLP-1 RAs’ Effectiveness in Atherosclerosis
The beneficial cardiovascular effect of GLP-1 RAs that has been demonstrated in CVOTs is mainly associated to the prevention of atherosclerotic events, especially in a decrease in non-fatal strokes and non-fatal myocardial infractions. These effects cannot be attributed only to the activity of GLP-1 RAs on controlling glucose levels and reducing body weight [134][92]. Multiple actions on inflammation, lipid levels, vascular smooth muscle cells (VSMC), and endothelium seem to be involved (Figure 2), with the exact mechanisms still under investigation [139][93]. A meta-analysis performed to examine the effects of GLP-1 RAs in atherosclerosis demonstrated their beneficial role through a decrease in the blood levels of plasminogen activator inhibitor-1 (PAI-1), high-sensitivity c-reactive protein (hsCRP), and brain natriuretic peptide (BNP), all of which are atherosclerosis markers, as well as total and LDL cholesterol and triglycerides [155][94]. A study on the cardio-metabolic effect of liraglutide in T2DM patients with MetS, with a total follow-up time of 18 weeks and no control group, showed that the drug (in combination with metformin) was able to decrease the carotid intima–media thickness (cIMT), a biomarker of subclinical atherosclerosis, as well as MetS prevalence, with a significant association between these two being identified. These results were seen early in the study (only 6 months after treatment initiation) [156][95]. Additionally, total and LDL cholesterol as well as triglycerides were found to be reduced during treatment [156][95], although a later post hoc analysis for the LEADER trial suggested that liraglutide’s benefits are not related to LDL-C levels since they were evident, even in the very low baseline LDL-C [157][96]. Recently, liraglutide was found to decrease the atherogenic small dense LDL-3 and LDL-4 subfractions of LDL cholesterol, with the former being correlated with a reduction in cIMT, which further enhances the theory that GLP-1 RAs’ effect is exerted separately from their glycemic and weight control effect [158][97]. A recent clinical study performed by Yang et al. (2021) separated the beneficial CV effect of GLP-1 RAs in terms of atherosclerosis progression from their hypoglycemic effect in patients with coronary heart disease (CHD), indicating that their role is associated with the polarization of macrophages towards M2, i.e., the anti-inflammatory type (Figure 2) [159][98]. The anti-inflammatory effect of liraglutide has also been previously reported from preclinical studies [160][99]. Specifically, liraglutide’s effect in pre-established atherosclerosis has been studied in vivo in ApoE−/− mice and ex vivo in human atherosclerotic plaques, with the results indicating the drug’s ability to reduce M1 proinflammatory mediators, such as MCP-1, TNF-a, and IL-1b, and upregulate the cathepsin protein family in the bone marrow, leading to an attenuation of atherosclerosis in the aorta through an increase in M2-like macrophages [160][99]. Changes in macrophages towards the M2 phenotype were also reported for exenatide based on in vitro experiments [161][100]. Liraglutide has also been found to improve plaque stability and hinder atherosclerotic plaque development in apolipoprotein-E-deficient (ApoE−/−) mice by suppressing endothelial dysfunction and the expression of vascular adhesion molecules (Figure 2) [162][101]. Similar results were obtained in another study performed by Rakipovsi et al. (2018) in ApoE−/− and low-density lipoprotein receptor-deficient (LDLr−/−) mice, demonstrating that liraglutide and semaglutide significantly lessened the development of plaque lesions, mainly due to anti-inflammatory effects. In this case, reductions in body weight and cholesterol levels were only partially involved, taking into consideration that the model used in that study was non-diabetic. These results were based on the observed reduction in the blood levels of systemic inflammation markers (TNF-a and interferon-γ) and the downregulation of various inflammatory pathways based on a transcription analysis of aortic atherosclerotic tissue (modified gene expression of proteins involved in leucocyte recruitment, adhesion, and migration) [163][102]. Additionally, liraglutide administered in ApoE−/− mice was found to exert its protective role mainly through down-regulating ACAT1 (responsible for producing cholesterol ester from free cholesterol), accompanied by the upregulation of ATP-binding cassette transporter A1 (ABCA1) (responsible for the efflux of free cholesterol) and the downregulation of scavenger receptor CD36 (responsible for uptake of oxLDL), which are involved in the monocyte/macrophage infiltration in the walls of arteries and the transformation of macrophages into foam cells [164][103]. Plaque stability has also been linked with the ability of GLP-1 RAs to modify the levels of matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs). MMPs can degrade the extracellular matrix, leading to intima thickening and vascular remodeling, and are modulated by TIMPs. Exenatide was found to regulate the in vitro expression of both MMPs and TIMPs in human coronary artery and aortic endothelial cells as well as coronary artery smooth muscle cells by suppressing NF-κB and Akt-Thr308 phosphorylation [165,166][104][105]. Similar results were obtained for liraglutide in C57BL/6J mice, with a reduced expression of MMP-9 as well as MCP-1 and ICAM-1, which were attributed to the exerted regulation of the Akt and extracellular signal-regulated kinase (ERK) pathways [167][106]. Additionally, exenatide was found to decrease oxidative stress (by suppressing the expression of NADPH oxidase, which is involved in ROS production) and ameliorate antioxidative potential (by enhancing the expression and activity of antioxidative enzymes SOD and GSH-Px) in human macrophages, processes that are involved in the pathogenesis and acceleration of atherosclerosis [168][107]. The protective role of GLP-1 RAs, specifically dulaglutide, in the endothelium regarding atherosclerosis was demonstrated in preclinical studies in human aortic endothelial cells (HAECs). Dulaglutide was found to prevent the atherosclerotic effects induced by oxLDL by preventing p53 protein phosphorylation and thus averting the downregulation of Krüppel-like Factor 2 (KLF2), a factor that greatly contributes to vascular endothelial cell protection by inhibiting monocyte adhesion to the endothelium and promoting nitric oxide synthase expression by endothelial cells (eNOS). Hence, dulaglutide hindered the production of proinflammatory cytokines and chemokines (such as IL-1β, IL-6, MCP-1, and high-mobility group protein 1 (HMGB-1)) as well as molecules that induce the adhesion of monocytes to endothelial cells, especially VCAM-1 and E-selectin [169][108]. Similar observations were made for luraglutide on KLF2, where its protective effect was found to be dependent on ERK-5 [170][109]. Exenatide was also found to decrease the recruitment of macrophages from the circulation and adhesion to the vessel wall [152][89]. The direct protective anti-inflammatory effect of GLP-1 RAs on the endothelium has also been elucidated from in vivo experiments in non-diabetic hypertensive mice, where liraglutide was able to decrease vascular inflammation and oxidative stress by averting endothelial NO synthetase (eNOS) uncoupling (thus inhibiting the production of ROS) and enhancing NO bioavailability, actions that are related to GLP-1 receptors in the endothelial cells but not on myeloid cells (Figure 2) [171][110]. Furthermore, liraglutide was found to obstruct platelet activation though in vitro experiments, a process that is involved in the pathogenesis of atherosclerosis by activating pathways that lead to NO production [172][111]. The protective action of GLP1-RAs on the endothelium, as well as their ability to reduce oxidative stress and, hence, endothelium disfunction and autophagy, has also been reported by other groups [173,174,175][112][113][114].References
- European Association for the Study of the Liver; European Association for the Study of Diabetes; European Association for the Study of Obesity. EASL-EASD-EASO Clinical Practice Guidelines for the Management of Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2016, 64, 1388–1402.
- Gluvic, Z.; Tomasevic, R.; Bojovic, K.; Obradovic, M.; Isenovic, E.R. Non-Alcoholic Fatty Liver Disease: A Multidisciplinary Clinical Practice Approach—the Institutional Adaptation to Existing Clinical Practice Guidelines. Emerg. Crit. Care Med. 2022, 2, 12–22.
- Naeem, M.; Markus, M.R.P.; Mousa, M.; Schipf, S.; Dörr, M.; Steveling, A.; Aghdassi, A.; Kühn, J.-P.; Kromrey, M.-L.; Nauck, M.; et al. Associations of Liver Volume and Other Markers of Hepatic Steatosis with All-Cause Mortality in the General Population. Liver Int. 2022, 42, 575–584.
- Choi, K.M.; Han, K.; Park, S.; Chung, H.S.; Kim, N.H.; Yoo, H.J.; Seo, J.-A.; Kim, S.G.; Kim, N.H.; Baik, S.H.; et al. Implication of Liver Enzymes on Incident Cardiovascular Diseases and Mortality: A Nationwide Population-Based Cohort Study. Sci. Rep. 2018, 8, 3764.
- Karaphillis, E.; Goldstein, R.; Murphy, S.; Qayyum, R. Serum Alanine Aminotransferase Levels and All-Cause Mortality. Eur. J. Gastroenterol. Hepatol. 2017, 29, 284–288.
- Kim, K.-N.; Joo, J.; Sung, H.K.; Kim, C.H.; Kim, H.; Kwon, Y.J. Associations of Serum Liver Enzyme Levels and Their Changes over Time with All-Cause and Cause-Specific Mortality in the General Population: A Large-Scale National Health Screening Cohort Study. BMJ Open 2019, 9, e026965.
- Noureddin, M.; Lam, J.; Peterson, M.R.; Middleton, M.; Hamilton, G.; Le, T.-A.; Bettencourt, R.; Changchien, C.; Brenner, D.A.; Sirlin, C.; et al. Utility of Magnetic Resonance Imaging versus Histology for Quantifying Changes in Liver Fat in Nonalcoholic Fatty Liver Disease Trials. Hepatology 2013, 58, 1930–1940.
- Loomba, R.; Neuschwander-Tetri, B.A.; Sanyal, A.; Chalasani, N.; Diehl, A.M.; Terrault, N.; Kowdley, K.; Dasarathy, S.; Kleiner, D.; Behling, C.; et al. Multicenter Validation of Association Between Decline in MRI-PDFF and Histologic Response in NASH. Hepatology 2020, 72, 1219–1229.
- Ajmera, V.; Loomba, R. Imaging Biomarkers of NAFLD, NASH, and Fibrosis. Mol. Metab. 2021, 50, 101167.
- Hernaez, R.; Lazo, M.; Bonekamp, S.; Kamel, I.; Brancati, F.L.; Guallar, E.; Clark, J.M. Diagnostic Accuracy and Reliability of Ultrasonography for the Detection of Fatty Liver: A Meta-Analysis. Hepatology 2011, 54, 1082–1090.
- Meroni, M.; Longo, M.; Lombardi, R.; Paolini, E.; Macchi, C.; Corsini, A.; Sirtori, C.R.; Fracanzani, A.L.; Ruscica, M.; Dongiovanni, P. Low Lipoprotein(a) Levels Predict Hepatic Fibrosis in Patients with Nonalcoholic Fatty Liver Disease. Hepatol. Commun. 2022, 6, 535–549.
- Papatheodoridi, M.; Cholongitas, E. Diagnosis of Non-Alcoholic Fatty Liver Disease (NAFLD): Current Concepts. Curr. Pharm. Des. 2018, 24, 4574–4586.
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and Validation of a Histological Scoring System for Nonalcoholic Fatty Liver Disease. Hepatology 2005, 41, 1313–1321.
- Brunt, E.M.; Kleiner, D.E.; Wilson, L.A.; Belt, P.; Neuschwander-Tetri, B.A.; NASH Clinical Research Network (CRN). Nonalcoholic Fatty Liver Disease (NAFLD) Activity Score and the Histopathologic Diagnosis in NAFLD: Distinct Clinicopathologic Meanings. Hepatology 2011, 53, 810–820.
- Manne, V.; Handa, P.; Kowdley, K.V. Pathophysiology of Nonalcoholic Fatty Liver Disease/Nonalcoholic Steatohepatitis. Clin. Liver Dis. 2018, 22, 23–37.
- Gerges, S.H.; Wahdan, S.A.; Elsherbiny, D.A.; El-Demerdash, E. Non-Alcoholic Fatty Liver Disease: An Overview of Risk Factors, Pathophysiological Mechanisms, Diagnostic Procedures, and Therapeutic Interventions. Life Sci. 2021, 271, 119220.
- Leamy, A.K.; Egnatchik, R.A.; Young, J.D. Molecular Mechanisms and the Role of Saturated Fatty Acids in the Progression of Non-Alcoholic Fatty Liver Disease. Prog. Lipid Res. 2013, 52, 165–174.
- Jensen, T.; Abdelmalek, M.F.; Sullivan, S.; Nadeau, K.J.; Green, M.; Roncal, C.; Nakagawa, T.; Kuwabara, M.; Sato, Y.; Kang, D.-H.; et al. Fructose and Sugar: A Major Mediator of Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2018, 68, 1063–1075.
- Kořínková, L.; Pražienková, V.; Černá, L.; Karnošová, A.; Železná, B.; Kuneš, J.; Maletínská, L. Pathophysiology of NAFLD and NASH in Experimental Models: The Role of Food Intake Regulating Peptides. Front. Endocrinol. 2020, 11, 597583.
- Chung, M.; Ma, J.; Patel, K.; Berger, S.; Lau, J.; Lichtenstein, A.H. Fructose, High-Fructose Corn Syrup, Sucrose, and Nonalcoholic Fatty Liver Disease or Indexes of Liver Health: A Systematic Review and Meta-Analysis. Am. J. Clin. Nutr. 2014, 100, 833–849.
- Zhang, X.; Ji, X.; Wang, Q.; Li, J.Z. New Insight into Inter-Organ Crosstalk Contributing to the Pathogenesis of Non-Alcoholic Fatty Liver Disease (NAFLD). Protein Cell 2018, 9, 164–177.
- Schaap, F.G.; Trauner, M.; Jansen, P.L.M. Bile Acid Receptors as Targets for Drug Development. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 55–67.
- Inagaki, T.; Moschetta, A.; Lee, Y.-K.; Peng, L.; Zhao, G.; Downes, M.; Yu, R.T.; Shelton, J.M.; Richardson, J.A.; Repa, J.J.; et al. Regulation of Antibacterial Defense in the Small Intestine by the Nuclear Bile Acid Receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 3920–3925.
- Ridlon, J.M.; Kang, D.-J.; Hylemon, P.B. Bile Salt Biotransformations by Human Intestinal Bacteria. J. Lipid Res. 2006, 47, 241–259.
- Puri, P.; Sanyal, A.J. The Intestinal Microbiome in Nonalcoholic Fatty Liver Disease. Clin. Liver Dis. 2018, 22, 121–132.
- Molinero, N.; Ruiz, L.; Sánchez, B.; Margolles, A.; Delgado, S. Intestinal Bacteria Interplay with Bile and Cholesterol Metabolism: Implications on Host Physiology. Front. Physiol. 2019, 10, 185.
- Buechler, C.; Wanninger, J.; Neumeier, M. Adiponectin, a Key Adipokine in Obesity Related Liver Diseases. World J. Gastroenterol. 2011, 17, 2801–2811.
- Boutari, C.; Perakakis, N.; Mantzoros, C.S. Association of Adipokines with Development and Progression of Nonalcoholic Fatty Liver Disease. Endocrinol. Metab. 2018, 33, 33–43.
- Polyzos, S.A.; Aronis, K.N.; Kountouras, J.; Raptis, D.D.; Vasiloglou, M.F.; Mantzoros, C.S. Circulating Leptin in Non-Alcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Diabetologia 2016, 59, 30–43.
- Targher, G.; Byrne, C.D.; Lonardo, A.; Zoppini, G.; Barbui, C. Non-Alcoholic Fatty Liver Disease and Risk of Incident Cardiovascular Disease: A Meta-Analysis. J. Hepatol. 2016, 65, 589–600.
- Sanders, F.W.B.; Griffin, J.L. De Novo Lipogenesis in the Liver in Health and Disease: More than Just a Shunting Yard for Glucose. Biol. Rev. Camb. Philos. Soc. 2016, 91, 452–468.
- Bessone, F.; Razori, M.V.; Roma, M.G. Molecular Pathways of Nonalcoholic Fatty Liver Disease Development and Progression. Cell. Mol. Life Sci. 2019, 76, 99–128.
- Bugianesi, E.; Moscatiello, S.; Ciaravella, M.F.; Marchesini, G. Insulin Resistance in Nonalcoholic Fatty Liver Disease. Curr. Pharm. Des. 2010, 16, 1941–1951.
- Lambert, J.E.; Ramos-Roman, M.A.; Browning, J.D.; Parks, E.J. Increased de Novo Lipogenesis Is a Distinct Characteristic of Individuals with Nonalcoholic Fatty Liver Disease. Gastroenterology 2014, 146, 726–735.
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of Fatty Acids Stored in Liver and Secreted via Lipoproteins in Patients with Nonalcoholic Fatty Liver Disease. J. Clin. Investig. 2005, 115, 1343–1351.
- Cohen, J.C.; Horton, J.D.; Hobbs, H.H. Human Fatty Liver Disease: Old Questions and New Insights. Science 2011, 332, 1519–1523.
- Puri, P.; Baillie, R.A.; Wiest, M.M.; Mirshahi, F.; Choudhury, J.; Cheung, O.; Sargeant, C.; Contos, M.J.; Sanyal, A.J. A Lipidomic Analysis of Nonalcoholic Fatty Liver Disease. Hepatology 2007, 46, 1081–1090.
- Ioannou, G.N. The Role of Cholesterol in the Pathogenesis of NASH. Trends Endocrinol. Metab. 2016, 27, 84–95.
- Adams, L.A.; Sanderson, S.O.; Lindor, K.D.; Angulo, P. The Histological Course of Nonalcoholic Fatty Liver Disease: A Longitudinal Study of 103 Patients with Sequential Liver Biopsies. J. Hepatol. 2005, 42, 132–138.
- Brunt, E.M.; Neuschwander-Tetri, B.A.; Oliver, D.; Wehmeier, K.R.; Bacon, B.R. Nonalcoholic Steatohepatitis: Histologic Features and Clinical Correlations with 30 Blinded Biopsy Specimens. Hum. Pathol. 2004, 35, 1070–1082.
- Foroughi, M.; Maghsoudi, Z.; Khayyatzadeh, S.; Ghiasvand, R.; Askari, G.; Iraj, B. Relationship between Non-Alcoholic Fatty Liver Disease and Inflammation in Patients with Non-Alcoholic Fatty Liver. Adv. Biomed. Res. 2016, 5, 28.
- Grattagliano, I.; de Bari, O.; Bernardo, T.C.; Oliveira, P.J.; Wang, D.Q.-H.; Portincasa, P. Role of Mitochondria in Nonalcoholic Fatty Liver Disease-from Origin to Propagation. Clin. Biochem. 2012, 45, 610–618.
- Koliaki, C.C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.J.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T.; et al. Adaptation of Hepatic Mitochondrial Function in Humans with Non-Alcoholic Fatty Liver Is Lost in Steatohepatitis. Cell Metab. 2015, 21, 739–746.
- Begriche, K.; Massart, J.; Robin, M.-A.; Bonnet, F.; Fromenty, B. Mitochondrial Adaptations and Dysfunctions in Nonalcoholic Fatty Liver Disease. Hepatology 2013, 58, 1497–1507.
- Caligiuri, A.; Gentilini, A.; Marra, F. Molecular Pathogenesis of NASH. Int. J. Mol. Sci. 2016, 17, 1575.
- Tilg, H.; Moschen, A.R. Evolution of Inflammation in Nonalcoholic Fatty Liver Disease: The Multiple Parallel Hits Hypothesis. Hepatology 2010, 52, 1836–1846.
- Schuster, S.; Cabrera, D.; Arrese, M.; Feldstein, A.E. Triggering and Resolution of Inflammation in NASH. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 349–364.
- Canbay, A.; Feldstein, A.E.; Higuchi, H.; Werneburg, N.; Grambihler, A.; Bronk, S.F.; Gores, G.J. Kupffer Cell Engulfment of Apoptotic Bodies Stimulates Death Ligand and Cytokine Expression. Hepatology 2003, 38, 1188–1198.
- Sutti, S.; Albano, E. Adaptive Immunity: An Emerging Player in the Progression of NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 81–92.
- Wree, A.; McGeough, M.D.; Peña, C.A.; Schlattjan, M.; Li, H.; Inzaugarat, M.E.; Messer, K.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. NLRP3 Inflammasome Activation Is Required for Fibrosis Development in NAFLD. J. Mol. Med. 2014, 92, 1069–1082.
- Boaru, S.G.; Borkham-Kamphorst, E.; Tihaa, L.; Haas, U.; Weiskirchen, R. Expression Analysis of Inflammasomes in Experimental Models of Inflammatory and Fibrotic Liver Disease. J. Inflamm. 2012, 9, 49.
- Friedman, S.L. Hepatic Stellate Cells: Protean, Multifunctional, and Enigmatic Cells of the Liver. Physiol. Rev. 2008, 88, 125–172.
- Seki, E.; Schwabe, R.F. Hepatic Inflammation and Fibrosis: Functional Links and Key Pathways. Hepatology 2015, 61, 1066–1079.
- Murphy, F.R.; Issa, R.; Zhou, X.; Ratnarajah, S.; Arthur, M.J.P.; Benyon, C.; Iredale, J.P.; Nagase, H. Inhibition of Apoptosis of Activated Hepatic Stellate Cells by Tissue Inhibitor of Metalloproteinase-1 Is Mediated via Effects on Matrix Metalloproteinase Inhibition: Implications for reversibility of liver fibrosis. J. Biol. Chem. 2002, 277, 11069–11076.
- Mantovani, A.; Ballestri, S.; Lonardo, A.; Targher, G. Cardiovascular Disease and Myocardial Abnormalities in Nonalcoholic Fatty Liver Disease. Dig. Dis. Sci. 2016, 61, 1246–1267.
- Mantovani, A.; Csermely, A.; Petracca, G.; Beatrice, G.; Corey, K.E.; Simon, T.G.; Byrne, C.D.; Targher, G. Non-Alcoholic Fatty Liver Disease and Risk of Fatal and Non-Fatal Cardiovascular Events: An Updated Systematic Review and Meta-Analysis. Lancet Gastroenterol. Hepatol. 2021, 6, 903–913.
- Byrne, C.D.; Targher, G. Non-Alcoholic Fatty Liver Disease Is a Risk Factor for Cardiovascular and Cardiac Diseases: Further Evidence That a Holistic Approach to Treatment Is Needed. Gut 2021.
- Kim, B.J.; Kim, N.H.; Kim, B.S.; Kang, J.H. The Association between Nonalcoholic Fatty Liver Disease, Metabolic Syndrome and Arterial Stiffness in Nondiabetic, Nonhypertensive Individuals. Cardiology 2012, 123, 54–61.
- Gómez, M.; Vila, J.; Elosua, R.; Molina, L.; Bruguera, J.; Sala, J.; Masià, R.; Covas, M.I.; Marrugat, J.; Fitó, M. Relationship of Lipid Oxidation with Subclinical Atherosclerosis and 10-Year Coronary Events in General Population. Atherosclerosis 2014, 232, 134–140.
- Sookoian, S.; Gianotti, T.F.; Rosselli, M.S.; Burgueño, A.L.; Castaño, G.O.; Pirola, C.J. Liver Transcriptional Profile of Atherosclerosis-Related Genes in Human Nonalcoholic Fatty Liver Disease. Atherosclerosis 2011, 218, 378–385.
- Lonardo, A.; Sookoian, S.; Pirola, C.J.; Targher, G. Non-Alcoholic Fatty Liver Disease and Risk of Cardiovascular Disease. Metabolism 2016, 65, 1136–1150.
- Lee, H.-H.; Cho, Y.; Choi, Y.J.; Huh, B.W.; Lee, B.-W.; Kang, E.S.; Park, S.W.; Cha, B.-S.; Lee, E.J.; Lee, Y.; et al. Non-Alcoholic Steatohepatitis and Progression of Carotid Atherosclerosis in Patients with Type 2 Diabetes: A Korean Cohort Study. Cardiovasc. Diabetol. 2020, 19, 81.
- Oni, E.; Budoff, M.J.; Zeb, I.; Li, D.; Veledar, E.; Polak, J.F.; Blankstein, R.; Wong, N.D.; Blaha, M.J.; Agatston, A.; et al. Nonalcoholic Fatty Liver Disease Is Associated With Arterial Distensibility and Carotid Intima-Media Thickness: (From the Multi-Ethnic Study of Atherosclerosis). Am. J. Cardiol. 2019, 124, 534–538.
- Madan, S.A.; John, F.; Pyrsopoulos, N.; Pitchumoni, C.S. Nonalcoholic Fatty Liver Disease and Carotid Artery Atherosclerosis in Children and Adults: A Meta-Analysis. Eur. J. Gastroenterol. Hepatol. 2015, 27, 1237–1248.
- Sookoian, S.; Pirola, C.J. Non-Alcoholic Fatty Liver Disease Is Strongly Associated with Carotid Atherosclerosis: A Systematic Review. J. Hepatol. 2008, 49, 600–607.
- Rampally, V.; Biri, S.K.; Nair, I.K.; Vadlakonda, A. Determination of Association between Nonalcoholic Fatty Liver Disease and Carotid Artery Atherosclerosis among Nondiabetic Individuals. J. Fam. Med. Prim. Care 2020, 9, 1182–1186.
- Guo, Y.-C.; Zhou, Y.; Gao, X.; Yao, Y.; Geng, B.; Cui, Q.-H.; Yang, J.-C.; Hu, H.-P. Association between Nonalcoholic Fatty Liver Disease and Carotid Artery Disease in a Community-Based Chinese Population: A Cross-Sectional Study. Chin. Med. J. 2018, 131, 2269–2276.
- Oni, E.T.; Agatston, A.S.; Blaha, M.J.; Fialkow, J.; Cury, R.; Sposito, A.; Erbel, R.; Blankstein, R.; Feldman, T.; Al-Mallah, M.H.; et al. A Systematic Review: Burden and Severity of Subclinical Cardiovascular Disease among Those with Nonalcoholic Fatty Liver; Should We Care? Atherosclerosis 2013, 230, 258–267.
- Orekhov, A.N.; Ivanova, E.A. Introduction of the Special Issue “Atherosclerosis and Related Diseases”. Vessel Plus 2017, 1, 163–165.
- Glass, C.K.; Witztum, J.L. Atherosclerosis: The Road Ahead. Cell 2001, 104, 503–516.
- RIDKER, P.M. Testing the Inflammatory Hypothesis of Atherothrombosis: Scientific Rationale for the Cardiovascular Inflammation Reduction Trial (CIRT). J. Thromb. Haemost. 2009, 7, 332–339.
- Libby, P. Inflammation in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2045–2051.
- Lind, L. Circulating Markers of Inflammation and Atherosclerosis. Atherosclerosis 2003, 169, 203–214.
- Patel, S.; Celermajer, D.S.; Bao, S. Atherosclerosis—Underlying Inflammatory Mechanisms and Clinical Implications. Int. J. Biochem. Cell Biol. 2008, 40, 576–580.
- Malekmohammad, K.; Sewell, R.D.E.; Rafieian-Kopaei, M. Antioxidants and Atherosclerosis: Mechanistic Aspects. Biomolecules 2019, 9, 301.
- Saponaro, C.; Gaggini, M.; Carli, F.; Gastaldelli, A. The Subtle Balance between Lipolysis and Lipogenesis: A Critical Point in Metabolic Homeostasis. Nutrients 2015, 7, 9453–9474.
- Singh, I.M.; Shishehbor, M.H.; Ansell, B.J. High-Density Lipoprotein as a Therapeutic TargetA Systematic Review. JAMA 2007, 298, 786–798.
- Bieghs, V.; Rensen, P.C.N.; Hofker, M.H.; Shiri-Sverdlov, R. NASH and Atherosclerosis Are Two Aspects of a Shared Disease: Central Role for Macrophages. Atherosclerosis 2012, 220, 287–293.
- Gustafson, B.; Hammarstedt, A.; Andersson, C.X.; Smith, U. Inflamed Adipose Tissue. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2276–2283.
- Bonetti, P.O.; Lerman, L.O.; Lerman, A. Endothelial Dysfunction. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 168–175.
- Lechner, K.; McKenzie, A.L.; Kränkel, N.; Von Schacky, C.; Worm, N.; Nixdorff, U.; Lechner, B.; Scherr, J.; Weingärtner, O.; Krauss, R.M. High-Risk Atherosclerosis and Metabolic Phenotype: The Roles of Ectopic Adiposity, Atherogenic Dyslipidemia, and Inflammation. Metab. Syndr. Relat. Disord. 2020, 18, 176–185.
- Nauck, M.A.; Quast, D.R.; Wefers, J.; Meier, J.J. GLP-1 Receptor Agonists in the Treatment of Type 2 Diabetes–State-of-the-Art. Mol. Metab. 2021, 46, 101102.
- Jendle, J.; Nauck, M.A.; Matthews, D.R.; Frid, A.; Hermansen, K.; Düring, M.; Zdravkovic, M.; Strauss, B.J.; Garber, A.J.; LEAD-2 and LEAD-3 Study Groups. Weight Loss with Liraglutide, a Once-Daily Human Glucagon-like Peptide-1 Analogue for Type 2 Diabetes Treatment as Monotherapy or Added to Metformin, Is Primarily as a Result of a Reduction in Fat Tissue. Diabetes Obes. Metab. 2009, 11, 1163–1172.
- Shiomi, M.; Tanaka, Y.; Takada, T.; Otori, K. Determining Whether the Effect of Liraglutide on Non-Alcoholic Fatty Liver Disease Depends on Reductions in the Body Mass Index. JGH Open 2020, 4, 995–1001.
- Smati, S.; Canivet, C.M.; Boursier, J.; Cariou, B. Anti-Diabetic Drugs and NASH: From Current Options to Promising Perspectives. Expert Opin. Investig. Drugs 2021, 30, 813–825.
- Armstrong, M.J.; Hull, D.; Guo, K.; Barton, D.; Hazlehurst, J.M.; Gathercole, L.L.; Nasiri, M.; Yu, J.; Gough, S.C.; Newsome, P.N.; et al. Glucagon-like Peptide 1 Decreases Lipotoxicity in Non-Alcoholic Steatohepatitis. J. Hepatol. 2016, 64, 399–408.
- Yu, X.; Hao, M.; Liu, Y.; Ma, X.; Lin, W.; Xu, Q.; Zhou, H.; Shao, N.; Kuang, H. Liraglutide Ameliorates Non-Alcoholic Steatohepatitis by Inhibiting NLRP3 Inflammasome and Pyroptosis Activation via Mitophagy. Eur. J. Pharmacol. 2019, 864, 172715.
- Kalogirou, M.; Sinakos, E. Treating Nonalcoholic Steatohepatitis with Antidiabetic Drugs: Will GLP-1 Agonists End the Struggle? World J. Hepatol. 2018, 10, 790–794.
- Wang, Y.; Parlevliet, E.T.; Geerling, J.J.; van der Tuin, S.J.L.; Zhang, H.; Bieghs, V.; Jawad, A.H.M.; Shiri-Sverdlov, R.; Bot, I.; de Jager, S.C.A.; et al. Exendin-4 Decreases Liver Inflammation and Atherosclerosis Development Simultaneously by Reducing Macrophage Infiltration. Br. J. Pharmacol. 2014, 171, 723–734.
- Ghazanfar, H.; Kandhi, S.D.; Nawaz, I.; Javed, N.; Abraham, M.C.; Farag, M.; Mahasamudram, J.; Patel, V.B.; Altaf, F.; Patel, H. Role of Glucagon-Like Peptide-1 Receptor Agonists in the Management of Non-Alcoholic Steatohepatitis: A Clinical Review Article. Cureus 2021, 13, e15141.
- Wang, X.-C.; Gusdon, A.M.; Liu, H.; Qu, S. Effects of Glucagon-like Peptide-1 Receptor Agonists on Non-Alcoholic Fatty Liver Disease and Inflammation. World J. Gastroenterol. 2014, 20, 14821–14830.
- Gerstein, H.C.; Colhoun, H.M.; Dagenais, G.R.; Diaz, R.; Lakshmanan, M.; Pais, P.; Probstfield, J.; Riesmeyer, J.S.; Riddle, M.C.; Rydén, L.; et al. Dulaglutide and Cardiovascular Outcomes in Type 2 Diabetes (REWIND): A Double-Blind, Randomised Placebo-Controlled Trial. Lancet 2019, 394, 121–130.
- Zelniker, T.A.; Wiviott, S.D.; Raz, I.; Im, K.; Goodrich, E.L.; Furtado, R.H.M.; Bonaca, M.P.; Mosenzon, O.; Kato, E.T.; Cahn, A.; et al. Comparison of the Effects of Glucagon-Like Peptide Receptor Agonists and Sodium-Glucose Cotransporter 2 Inhibitors for Prevention of Major Adverse Cardiovascular and Renal Outcomes in Type 2 Diabetes Mellitus. Circulation 2019, 139, 2022–2031.
- Song, X.; Jia, H.; Jiang, Y.; Wang, L.; Zhang, Y.; Mu, Y.; Liu, Y. Anti-Atherosclerotic Effects of the Glucagon-like Peptide-1 (GLP-1) Based Therapies in Patients with Type 2 Diabetes Mellitus: A Meta-Analysis. Sci. Rep. 2015, 5, 10202.
- Rizzo, M.; Rizvi, A.A.; Patti, A.M.; Nikolic, D.; Giglio, R.V.; Castellino, G.; Li Volti, G.; Caprio, M.; Montalto, G.; Provenzano, V.; et al. Liraglutide Improves Metabolic Parameters and Carotid Intima-Media Thickness in Diabetic Patients with the Metabolic Syndrome: An 18-Month Prospective Study. Cardiovasc. Diabetol. 2016, 15, 162.
- Verma, S.; Leiter, L.A.; Mazer, C.D.; Bain, S.C.; Buse, J.; Marso, S.; Nauck, M.; Zinman, B.; Bosch-Traberg, H.; Rasmussen, S.; et al. Liraglutide Reduces Cardiovascular Events and Mortality in Type 2 Diabetes Mellitus Independently of Baseline Low-Density Lipoprotein Cholesterol Levels and Statin Use. Circulation 2018, 138, 1605–1607.
- Nikolic, D.; Giglio, R.V.; Rizvi, A.A.; Patti, A.M.; Montalto, G.; Maranta, F.; Cianflone, D.; Stoian, A.P.; Rizzo, M. Liraglutide Reduces Carotid Intima-Media Thickness by Reducing Small Dense Low-Density Lipoproteins in a Real-World Setting of Patients with Type 2 Diabetes: A Novel Anti-Atherogenic Effect. Diabetes Ther. 2021, 12, 261–274.
- Yang, L.; Chen, L.; Li, D.; Xu, H.; Chen, J.; Min, X.; He, M.; Wu, T.; Zhong, J.; Yang, H.; et al. Effect of GLP-1/GLP-1R on the Polarization of Macrophages in the Occurrence and Development of Atherosclerosis. Mediators Inflamm. 2021, 2021, 5568159.
- Bruen, R.; Curley, S.; Kajani, S.; Lynch, G.; O’Reilly, M.E.; Dillon, E.T.; Brennan, E.P.; Barry, M.; Sheehan, S.; McGillicuddy, F.C.; et al. Liraglutide Attenuates Preestablished Atherosclerosis in Apolipoprotein E–Deficient Mice via Regulation of Immune Cell Phenotypes and Proinflammatory Mediators. J. Pharmacol. Exp. Ther. 2019, 370, 447–458.
- Shiraishi, D.; Fujiwara, Y.; Komohara, Y.; Mizuta, H.; Takeya, M. Glucagon-like Peptide-1 (GLP-1) Induces M2 Polarization of Human Macrophages via STAT3 Activation. Biochem. Biophys. Res. Commun. 2012, 425, 304–308.
- Gaspari, T.; Welungoda, I.; Widdop, R.E.; Simpson, R.W.; Dear, A.E. The GLP-1 Receptor Agonist Liraglutide Inhibits Progression of Vascular Disease via Effects on Atherogenesis, Plaque Stability and Endothelial Function in an ApoE−/− Mouse Model. Diabetes Vasc. Dis. Res. 2013, 10, 353–360.
- Rakipovski, G.; Rolin, B.; Nøhr, J.; Klewe, I.; Frederiksen, K.S.; Augustin, R.; Hecksher-Sørensen, J.; Ingvorsen, C.; Polex-Wolf, J.; Knudsen, L.B. The GLP-1 Analogs Liraglutide and Semaglutide Reduce Atherosclerosis in ApoE−/− and LDLr−/− Mice by a Mechanism That Includes Inflammatory Pathways. JACC Basic to Transl. Sci. 2018, 3, 844–857.
- Tashiro, Y.; Sato, K.; Watanabe, T.; Nohtomi, K.; Terasaki, M.; Nagashima, M.; Hirano, T. A Glucagon-like Peptide-1 Analog Liraglutide Suppresses Macrophage Foam Cell Formation and Atherosclerosis. Peptides 2014, 54, 19–26.
- Gallego-Colon, E.; Klych-Ratuszny, A.; Kosowska, A.; Garczorz, W.; Mohammad, M.R.; Wozniak, M.; Francuz, T. Exenatide Modulates Metalloproteinase Expression in Human Cardiac Smooth Muscle Cells via the Inhibition of Akt Signaling Pathway. Pharmacol. Rep. 2018, 70, 178–183.
- Garczorz, W.; Gallego-Colon, E.; Kosowska, A.; Kłych-Ratuszny, A.; Woźniak, M.; Marcol, W.; Niesner, K.J.; Francuz, T. Exenatide Exhibits Anti-Inflammatory Properties and Modulates Endothelial Response to Tumor Necrosis Factor α-Mediated Activation. Cardiovasc. Ther. 2018, 36, e12317.
- Kim, J.H.; Lee, G.Y.; Maeng, H.J.; Kim, H.; Bae, J.H.; Kim, K.M.; Lim, S. Effects of Glucagon-Like Peptide-1 Analogue and Fibroblast Growth Factor 21 Combination on the Atherosclerosis-Related Process in a Type 2 Diabetes Mouse Model. Endocrinol. Metab. 2021, 36, 157–170.
- Bułdak, Ł.; Łabuzek, K.; Bułdak, R.J.; Machnik, G.; Bołdys, A.; Okopień, B. Exenatide (a GLP-1 Agonist) Improves the Antioxidative Potential of in Vitro Cultured Human Monocytes/Macrophages. Naunyn. Schmiedebergs. Arch. Pharmacol. 2015, 388, 905–919.
- Chang, W.; Zhu, F.; Zheng, H.; Zhou, Z.; Miao, P.; Zhao, L.; Mao, Z. Glucagon-like Peptide-1 Receptor Agonist Dulaglutide Prevents Ox-LDL-Induced Adhesion of Monocytes to Human Endothelial Cells: An Implication in the Treatment of Atherosclerosis. Mol. Immunol. 2019, 116, 73–79.
- Yue, W.; Li, Y.; Ou, D.; Yang, Q. The GLP-1 Receptor Agonist Liraglutide Protects against Oxidized LDL-Induced Endothelial Inflammation and Dysfunction via KLF2. IUBMB Life 2019, 71, 1347–1354.
- Helmstädter, J.; Frenis, K.; Filippou, K.; Grill, A.; Dib, M.; Kalinovic, S.; Pawelke, F.; Kus, K.; Kröller-Schön, S.; Oelze, M.; et al. Endothelial GLP-1 (Glucagon-Like Peptide-1) Receptor Mediates Cardiovascular Protection by Liraglutide In Mice With Experimental Arterial Hypertension. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 145–158.
- Barale, C.; Buracco, S.; Cavalot, F.; Frascaroli, C.; Guerrasio, A.; Russo, I. Glucagon-like Peptide 1-Related Peptides Increase Nitric Oxide Effects to Reduce Platelet Activation. Thromb. Haemost. 2017, 117, 1115–1128.
- Cai, X.; She, M.; Xu, M.; Chen, H.; Li, J.; Chen, X.; Zheng, D.; Liu, J.; Chen, S.; Zhu, J.; et al. GLP-1 Treatment Protects Endothelial Cells from Oxidative Stress-Induced Autophagy and Endothelial Dysfunction. Int. J. Biol. Sci. 2018, 14, 1696–1708.
- Tang, S.; Zhang, Q.; Tang, H.; Wang, C.; Su, H.; Zhou, Q.; Wei, W.; Zhu, H.; Wang, Y. Effects of Glucagon-like Peptide-1 on Advanced Glycation Endproduct-Induced Aortic Endothelial Dysfunction in Streptozotocin-Induced Diabetic Rats: Possible Roles of Rho Kinase- and AMP Kinase-Mediated Nuclear Factor ΚB Signaling Pathways. Endocrine 2016, 53, 107–116.
- Wu, Y.-C.; Wang, W.-T.; Lee, S.-S.; Kuo, Y.-R.; Wang, Y.-C.; Yen, S.-J.; Lee, M.-Y.; Yeh, J.-L. Glucagon-Like Peptide-1 Receptor Agonist Attenuates Autophagy to Ameliorate Pulmonary Arterial Hypertension through Drp1/NOX- and Atg-5/Atg-7/Beclin-1/LC3β Pathways. Int. J. Mol. Sci. 2019, 20, 3435.
More