Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Małgorzata Kujawska and Version 2 by Camila Xu.
In 1987, bacteria were found to insert 32-nt (nucleotide) spacer sequences into 29-nt repeat sequences in CRISPR loci whenever they came into contact with phage DNA, leading to the discovery of the CRISPR-Cas system. The CRISPR-Cas-based technology can be employed as a new genome-editing tool in various organisms. The new gene-editing technique holds great promise for research and therapy of neurodegenerative diseases (NDs), such as Parkinson's disease (PD), for which there are currently no effective disease-modifying treatments.
- Parkinson’s
- CRISPR-Cas9
- gene therapy
- delivery
1. CRISPR-Cas
1.1. History
In 1987, bacteria were found to insert 32-nt (nucleotide) spacer sequences into 29-nt repeat sequences in CRISPR loci whenever they came into contact with phage DNA, leading to the discovery of the CRISPR-Cas system [1][21]. Similar repeating sequences were discovered in other E. coli strains: enterobacteria closely related to E. coli, and Shigella dysentery in the following years [2][22]. In 1993, Mojica and colleagues found the CRISPR repetitive sequence in archaea while researching the effects of salinity on the growth of Haloferax mediterranei. Although there was no similarity between these sequences and E.coli repeats, these researchers discovered a lengthy DNA sequence in the genome of these archaea that consisted of regulatory repeats [3][23]. In the CRISPR-Cas era, 2005 is regarded as a pivotal year because it was recognized that the spacer sequences were derived from phage genomes [4][24]. Together with the finding that Cas-gene encoded proteins with putative helicase and nuclease domains [5][6][7][25,26,27], and that CRISPR loci can be transcribed [8][28], It was recommended that CRISPR-cas is an adaptive system that may use antisense RNAs as a memory marker of past invasions [9][29]. In 2007, it was suggested that the CRISPR system could be used as an adaptive immune defense for bacteria and archaea against phage attacks. For example, adding or deleting spacer DNA homologous to phage DNA can alter the resistance of Streptococcus thermophilus to phage invasion [10][30]. In 2008, mature CRISPR RNAs (crRNAs) were determined to act as guides in a complex with Cas proteins in E. coli, preventing viral replication [11][31]. The CRISPR-Cas system’s DNA targeting activity was identified in the pathogen Staphylococcus epidermidis the same year. For nearly 20 years after their discovery, the function of these repeats remained unknown. Multiple direct repeats (DRs), short regulatory spaced repeats, and large clusters of tandem repeats have all been proposed as names for these repeats. Jansen and coworkers invented the word CRISPR, which has now gained acceptance among researchers since it reflects the structural properties of repeats [12][13][14][15][32,33,34,35].
1.2. CRISPR-Cas System
The classification of the CRISPR-Cas system is very challenging because there are no universal Cas proteins that could have served as phylogenetic markers. Consequently, the classification is based on many features, including the layout of Cas operons, signature Cas genes, and phylogenies of conserved Cas proteins [16][36]. There are two classes (Class 1 and Class 2), six types (I–VI), and 33 subtypes of CRISPR-Cas, according to a classification published in 2020 [17][37]. Multi-subunit effector complexes are seen in Class 1, while single protein effector modules are found in Class 2. Identifying two new types and several subtypes of the Class 2 CRISPR-Cas system resulted in more research and analysis of the system. The type VI systems, out of the two recently identified and defined CRISPR types, were the only ones that targeted RNA. In some circumstances, the class 2 systems have a unique feature in which the effector protein is also involved in processing pre-crRNA (CRISPR RNA) [18][38]. The CRISPR-Cas system’s two major classes, 1 and 2, have a solid basis of variation. The multi-subunit crRNA effector complex is classified as Class 1, while the single crRNA effector complex has been classified as Class 2. The Class 1 CRISPR-Cas system has been subdivided into types (I, III, and IV) and further into subtypes. Similarly, Class 2 is divided into three types: II, V, and VI, each further classified into multiple subtypes. The most widely used CRISP-Cas system is the type II CRISPR-Cas system which has been obtained from Streptococcus pyogens (SpCas9) [19][20][39,40]. The two main components of the CRISPR-Cas9 system are single guided RNA (sgRNA) and RNA guided Cas9 endonuclease [21][41]. There are two nuclease domains of Cas9, named RuvC and HNH, each breaking a single strand of targeted double-stranded DNA [22][42]. The RuvC domain cleaves the non-complimentary strand of dsDNA interacting with crRNA, while the HNH domain cuts the complementary strand [23][43]. A single-guide RNA (sgRNA) is a condensed form of crRNA and tracrRNA [24][44]. The Cas9 nuclease and sgRNA combine to form a Cas9 ribonucleoprotein (RNP) that can bind to and cleave the specific target in DNA [23][43].
Furthermore, the desired task of CRISPR-Cas9 systems is provided by the protospacer adjacent motif (PAM), which is an area inside an invading DNA that helps bacteria in differentiating pathogenic genetic information from its own [25][26][45,46]. If the spacer sequence is entirely identical to PAM, the CRISPR-Cas9 system will exclusively target plasmid or viral genetic materials by generating double-stranded (ds) DNA breaks in the invaded DNA [27][47]. As a result of these findings, researchers have determined that the CRISPR-Cas9 system can be employed as a new genome-editing tool in various organisms. It causes double-strand breaks (DSB), which can be fixed by either the homologous directed repair pathway (HDR) or error-prone non-homologous end junction (NHEJ) pathway, which are both endogenous self-healing processes [28][48]. NHEJ is more effective than HDR in most cases because it does not depend on a nearby homology donor and is also active for approximately 90% of the cell cycle [29][49]. NHEJ can integrate random insertion or deletion (indel) into the cleavage site, resulting in frameshift mutation or early termination codon in the open reading frame of the target gene so as to inactivate it [30][31][50,51]. However, HDR can introduce precise genomic changes at the target site using homologous DNA repair templates [32][33][52,53]. In addition, many sgRNAs targeting one or more genes can be used to create large deletions and knock out many genes at the same time [34][35][54,55].
2. Parkinson’s Disease
Parkinson’s disease (PD), after Alzheimer’s disease, is the second most common human neurodegenerative disease characterized by affected body movements [36][56]. PD is a heterogeneous neurodegenerative condition that affects an estimated 10 million people globally [37][57]. The progressive loss of dopaminergic neurons (DNs) in the substantia nigra pars compacta (SNpc) causes motor symptoms such as rest tremors, bradykinesia, and rigidity, which constitute the core of PD clinical characteristics [38][58]. This neuronal loss is followed by the appearance of cytoplasmic inclusions of Lewy bodies (LBs), which are primarily made of aggregates of misfolded α-synuclein protein and may spread in a prion-like way between synaptically interconnected areas [39][59] that has been supported in in vitro, in vivo and autopsy studies [40][60].
In addition, non-motor symptoms like cognitive decline, sleeping problems, depression, intestinal dysfunction, and anxiety are also becoming more commonly recognized as key factors in a patient’s standard of living and impairment [41][61]. PD prevalence rises with age (from 40–49 years up to people aged >80 years), and it is gender-dependent, with it being twice as common in males than in females [42][43][62,63]. The incidence rate of PD worldwide is increasing, and by 2040, the number of people suffering from the disease is expected to be close to 12 million, prompting some scholars to list it as a pandemic [44][45][64,65]. The majority of PD patients are classed as idiopathic, with approximately 10% having a proven monogenic cause (familial PD). Idiopathic PD’s etiology is unknown, but genetics, aging and environmental factors and their interactions have a role in the disease’s onset and development. Ninety common polymorphisms linked to the development of PD have been discovered in genome-wide studies [46][47][11,66], and the influence of genetic factors on the clinical heterogeneity and development of PD is still being investigated. Currently, the most common treatment for PD is symptomatic medication therapy. No mechanism-based treatment methods to prevent, regulate, or minimize the clinical signs of PD have been developed [48][49][67,68]. Additionally, the symptomatic therapeutic modalities used have many side effects. With the progression of the disease, the nonlinear pharmacodynamics of dopamine (DA) replacement therapy complicates the optimization of a treatment regimen [50][69]. A few cell replacement therapy researchers have demonstrated the feasibility of producing DNs from human embryonic stem cells (hESCs) and implanting these cells in animal PD models [51][52][70,71]. The early findings revealed that DA levels in the brains of experimental animals had increased [53][72]. However, this technique has several unresolved issues, including the possibility of immunologic response, brain tumors, ethical considerations, phenotype instability of hESC-derived DA neurons, and the need to assess the treatment’s effectiveness and safety in PD patients.
3. Application of CRISPR-Cas in PD
Based on the observation that α-synuclein accumulation in microglia induced severe neurodegeneration of DNs [54][55][73,74], vaccines against α-synuclein might be an effective treatment option. However, no research focused on this method has yet been published. New mechanistic studies are needed to better understand the pathogenesis of PD, in which environmental and genetic variables contribute to a range of aberrant metabolic pathways and incorrect interactions between different macromolecules. The CRISPR-Cas9 system—a revolutionary technology created in the last decade that allows for immediate and accurate genome editing in nearly any living species—seems to be a promising approach in PD also [56][57][75,76]. CRISPR-Cas9 offers the possibility to accelerate basic research, focusing on elucidating the pathogenicity of neurological diseases and leading to new therapies, according to several recent articles, mainly for PD [58][59][77,78]. CRISPR-Cas9 technology is more succinct, versatile, and cost-effective than other gene-editing methods, resulting in its increasing popularity [21][41]. The CRISPR-Cas9 system enables us to edit candidate genes (Table 1) to generate appropriate animal and cell line models, significantly improving theour understanding of the disease. In the future, it may become an important tool for effective and valuable gene therapy, which is considered to be a new therapeutic strategy for PD [60][79].
Table 1.
Genes implicated in the development of PD, their loci, proteins, functions, phenotypes, and neuropathology.
| Genes | Gene Locus | Alternative Names of the Gene | Proteins | Gene Function | Results of Gene Mutation | Onset of PD | ||
|---|---|---|---|---|---|---|---|---|
| PRKN | 6q26 | PARK2 | Parkin | Parkin is a 465-amino-acid cytosolic E3 ubiquitin ligase that participates in proteasome-mediated protein degradation. It damages misfolded and overproduced proteins, as well as ubiquitin. | The absence of LB, dopaminergic neuron apoptosis in the SN, and neurofibrillary | Early | [61][62][63][64][65][66] | [80,81,82,83,84,85] |
| SNCA | 4q22.1 | PARK 1/PARK 4 | α-synuclein | The SNCA gene produces a protein called -synuclein, widely distributed in neurons. Its function is unknown; however, it may be involved in regulating vesicular and dopamine neurotransmission. | The broad presence of LB throughout the brain and cerebral cortex, as well as neuronal destruction in the LC and SN | Early | [67][68][69] | [86,87,88] |
| PINK1 | 1p36.12 | PARK6 | PTEN induced putative kinase 1 | The mitochondrial function of this protein is to protect the mitochondria from the damaging effects of cellular oxidative stress. | The occurrence of LB in the reticular nuclei of the brainstem and neuronal loss in the SN pars compacta | Early | [70][71][72] | [89,90,91] |
| RAB39B | Xq28 | None | RAB proteins, like RAB39B | These are members of the GTPase family. RAB39B controls the movement of vesicles between membrane compartments. | Extensive dopaminergic neuron loss in SN and classical LB disorder | X-linked early-onset | [73][74][75] | [92,93,94] |
| D-J1 | 1p36.23 | PARK7 | DJ-1 | Several tissue and organs, including the brain, contain the DJ-1 protein. This protein acts as a chaperone molecule and prevents cells from oxidative stress. DJ-1 assists in the refolding of damaged proteins as well as the assembly of specific proteins into the right three-dimensional shape. | LB pathology | Early | [76][77][78][79] | [95,96,97,98] |
| LRRK2 | 12q12 | PARK8 | Leucine-rich repeat kinase 2 | The protein Roco family includes the component of the gene LRRK2. It is involved in cytoskeletal dynamics, autophagy, and vesicular transport. | Heterogeneous: degeneration of neurons in the SN and occurrence of LB in the brain; specific cases: Neurofibrillary tangle pathology, lack of LB, and neural nigral degeneration | Late | [80][81][82] | [99,100,101] |
PD, Parkinson’s disease; SNCA, Synuclein alpha; SN, substantia nigra; LB, Lewy body; LC, locus coeruleus; LRRK2, leucine-rich repeat kinase 2; PINK1, PTEN-induce kinase 1.
CRISPR-Cas9 technologies have been proposed to offer a number of genomic modifications in addition to site-directed gene editing. CRISPR interference (CRISPRi) and CRISPR activation (CRISPRa) technologies have also been used to regulate the expression of target genes by making precise base modifications with a catalytically dead nuclease (dCas9) [83][84][85][86][102,103,104,105]. In addition, they have been adapted as tools for gene location detection [87][106], epigenetic research [88][107], and even modified RNA targeting (Figure 1) [89][108].
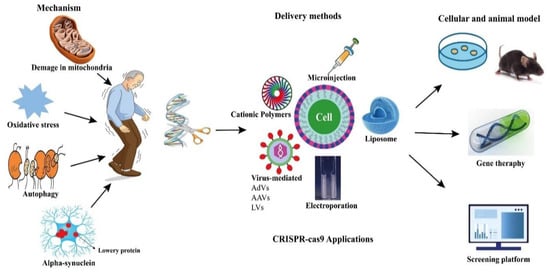
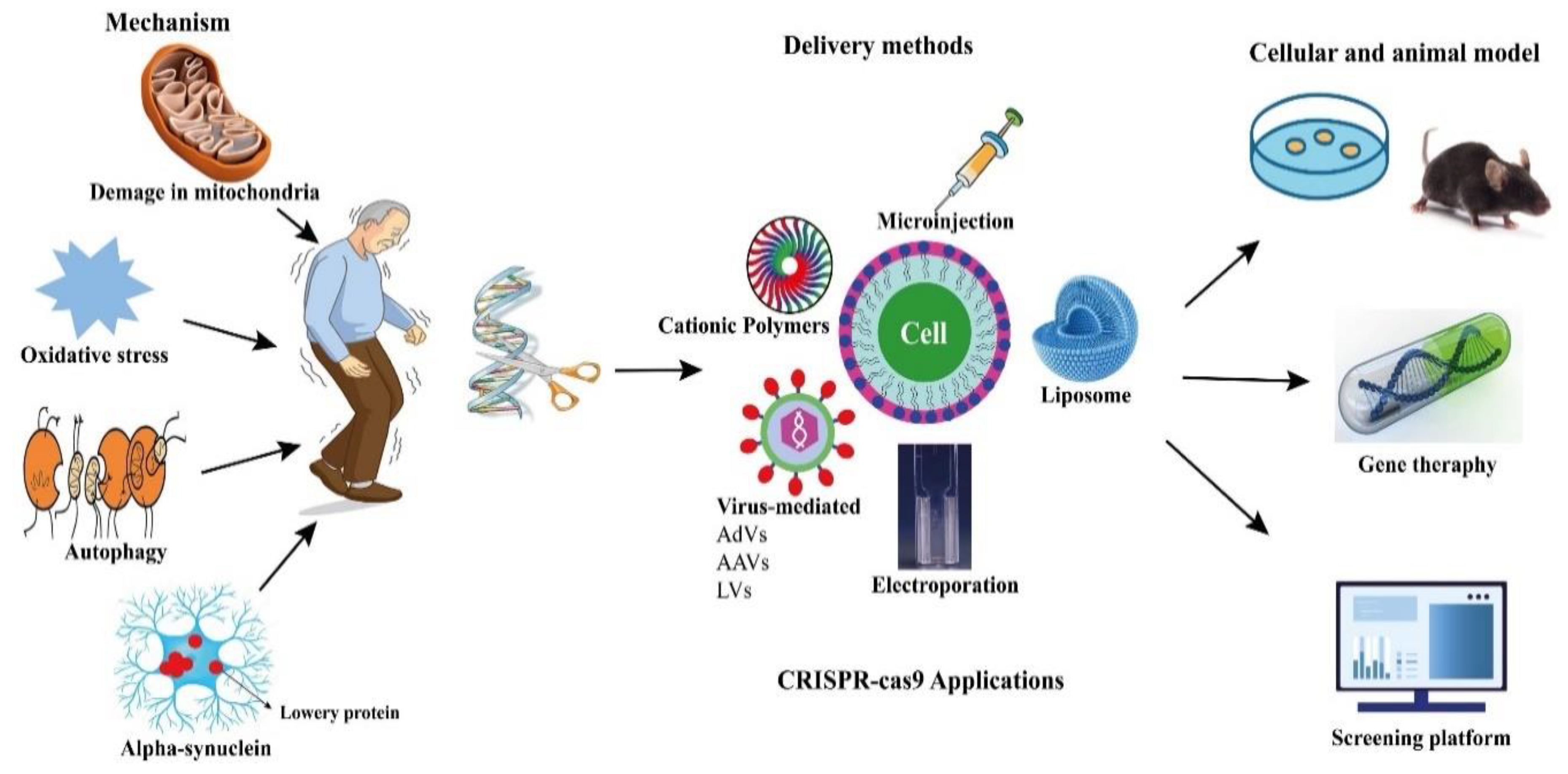
Figure 1. Potential applications of CRISPR-Cas9 in PD.
Potential applications of CRISPR-Cas9 in PD.