Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Eleftheria Galatou and Version 4 by Jessie Wu.
Nonalcoholic steatohepatitis (NASH) is the most severe manifestation of nonalcoholic fatty liver disease (NAFLD), a common complication of type 2 diabetes, and may lead to cirrhosis and hepatocellular carcinoma. Oxidative stress and liver cell damage are the major triggers of the severe hepatic inflammation that characterizes NASH, which is highly correlated with atherosclerosis and coronary artery disease.
- nonalcoholic steatohepatitis (NASH)
- nonalcoholic fatty liver disease (NAFLD)
- atherosclerosis
1. Diagnosis of Nonalcoholic Fatty Liver Disease/Nonalcoholic Steatohepatitis
NAFLD comprises a wide spectrum of liver diseases ranging from simple fat accumulation to more advanced stages, including NASH, cirrhosis, and cancer. The diagnosis of NAFLD requires the identification of hepatic steatosis (liver fat > 5%) in the absence of other causes of liver fat accumulation (e.g., alcohol consumption or coexisting causes of chronic liver diseases) [1][2][9,14]. NAFLD is often suspected in clinical practice when an individual presents features of MetS, such us abdominal obesity, hypertension, increased levels of triglycerides, low levels of HDL, and increased levels of fasting blood glucose. In the same context, individuals with persistently abnormal liver enzyme levels should be screened for NAFLD/NASH [1][9]. Several studies have shown that mild-to-moderate elevations in serum liver enzymes or increased liver volume [3][15] are associated with a higher risk of all-cause mortality [4][5][6][16,17,18]. There are many available imaging techniques, including ultrasound, image-guided biopsy, computed tomography (CT), and magnetic resonance imaging (MRI). Multiparametric MRI combines two or more quantitative techniques, such as T1, T2, and the proton density fat fraction (PDFF), to assess hepatic inflammation and fibrosis with a high level of accuracy. MRI-PDFF was more accurate at detecting changes in liver fat than liver biopsy and has been validated in multiple studies [7][8][9][19,20,21]. Ultrasound is widely accepted as a first-line diagnostic tool since it is a non-invasive, low cost, and radiation-free technique with a satisfactory sensitivity for moderate and severe steatosis identification [10][22]. To date, the gold standard to diagnose patients with NASH is still considered to be liver biopsy, demonstrating the typical fibrosis pattern, which cannot be seen via imaging methods. Currently, although there is no readily available, reliable, and non-invasive method to identify the progression of steatosis to NASH and fibrosis, altered levels of lipoprotein (a) (Lp(a)) [11][23] and several biomarkers of inflammation (such as ferritin and high-sensitivity C reactive protein (CRP)) and apoptosis (cytokeratine 18 (CK-18)) [12][24] have been associated with the diagnosis of NASH in NAFLD patients. Moreover, the NAFLD Activity Score (NAS) is used in clinical trials for evaluating the changes in histological features caused by therapeutic interventions. This system was developed and validated by the Pathology Committee of the NASH Clinical Research Network (NASH CRN) and is based on the semi-quantitative evaluation of histological features, specifically steatosis (0–3), lobular inflammation (0–2), and hepatocellular ballooning (0–2). NAS is calculated by adding these values, with a sum of ≥5 indicating NASH and scores < 3 considered as “not NASH” [13][25]. However, the threshold value of 5 in NAS is not always in accordance with a NASH diagnosis that is based on the analysis of a liver biopsy for the existence of certain lesions with specific patterns [14][26].
2. Pathophysiological Mechanisms of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis
NAFLD is a metabolic disorder, and its pathogenesis is a multifactorial process that involves a complex interaction between metabolic, clinical, environmental, and genetic factors [15][16][38,39].
2.1. Nutrition and Gut Microbiota
Components of nutrition and caloric intake play a key role in NAFLD development and progression. A nutritional high-fat and hypercaloric pattern rich in saturated fat and omega-6 (n-6) polyunsaturated fatty acids (PUFAs), carbohydrates, and low amounts of omega-3 (n-3) PUFAs and fibers have all been associated with NAFLD [17][40]. Data from preclinical and clinical studies demonstrate that fructose intake in hypercaloric diets is associated with an increased intrahepatic content of triglycerides, de novo lipogenesis, hepatic steatosis, obesity, and insulin resistance [18][19][41,42]. Glucose intake exerts similar detrimental effects on liver health by increasing hepatic lipid accumulation in healthy men [20][43]. This energy imbalance leads to higher post-prandial blood glucose levels and, thus, a higher insulin secretion rate. Insulin further stimulates de novo lipogenesis, leading to hepatic inflammation and subsequent NASH development. It has been demonstrated that a high-fat diet induces hepatic tumor necrosis factor-a (TNF-a) and interleukin (IL)-6 expression, whereas their inhibition prevents hepatic steatosis and NAFLD progression.
The body of evidence supporting an association between gut microbiota and NAFLD pathogenesis and progression is increasing. Basic players for complex dietary carbohydrate degradation are gut bacteria, leading to the production of short-chain fatty acid (SCFA) metabolites, including acetate, propionate, and butyrate. Although these metabolites improve glucose and lipid metabolism and maintain intestinal homeostasis, the increased production may contribute to obesity and liver steatosis, enhancing nutrient absorption [21][44]. Moreover, bile acids, which are synthesized by cholesterol within the liver, metabolized in the small intestine, and reabsorbed back to the liver through the portal vein, are significant regulators of lipid and carbohydrate metabolism and, subsequently, energy homeostasis. In the liver, they act as signaling molecules through their binding and activation of several nuclear hormone receptors, including farnesoid X receptors (FXRs) and G-protein-coupled receptor 5 (TGR5) [22][45]. Many studies have assessed the interplay between bile acids and microbiota, demonstrating that bile acids regulate and control the microbiome, while gut bacteria contribute to several biotransformations of bile acids and affect their composition, modulating hepatic steatosis [23][24][25][26][46,47,48,49].
It is well-documented that patients with biopsy-proven NAFLD/NASH exhibit a different microbiota signature and increased gut permeability due to a disruption of gut epithelial tight junctions. The leaky intestine leads to a translocation of bacteria and bacteria-produced endotoxins and alcohol through the portal circulation to the liver, contributing to ROS generation, hepatic inflammation through the toll-like receptor (TLR)-4-mediated pathway, and possibly fibrogenesis [26][27][28][49,50,51].
2.2. Adipose Tissue Dysfunction—The Role of Adipokines
Adipose tissue is an essential and highly active metabolic and endocrine organ that stores triacylglycerol as an energy source and releases adipokines and cytokines that are suspected to play a key role in NAFLD development and the progression to NASH. Visceral adiposity leads to excessive lipid accumulation and is highly correlated with insulin resistance due to an imbalance in pro-inflammatory and anti-inflammatory cytokine release. Leptin, an adipokine that plays a crucial role in the regulation of body weight and fat content, primarily acts centrally to reduce food intake, increase energy expenditure, and prevent lipid accumulation in organs other than the adipose tissue. However, excessive levels of leptin may result in hepatic inflammation and fibrosis [29][52]. Adiponectin, an abundant adipokine with anti-inflammatory and antifibrotic properties that acts on Kupffer cells and hepatic stellate cells (HSCs), increases hepatic insulin sensitivity by suppressing gluconeogenesis and lipogenesis and reduces body fat [27][50]. The anti-inflammatory effects of adiponectin are achieved by: (1) the suppression of transcription factor NF-κB, (2) the inhibition of pro-inflammatory cytokine release, and (3) the stimulation of anti-inflammatory cytokine secretion [28][51]. Patients with NAFLD and NASH exhibit elevated leptin levels and decreased adiponectin levels, which are associated with the severity of NAFLD patients, probably reflecting the increasing insulin resistance [29][30][8,52]. However, in later stages of NASH progression to cirrhosis, adiponectin levels are increased, possibly due to an impaired clearance of adiponectin and an excessive release of pro-inflammatory cytokines.
2.3. Insulin Resistance and Hepatic Fat Accumulation
Two pivotal characteristics of NAFLD pathophysiology are insulin resistance and hepatic steatosis. Insulin plays a crucial role in the regulation of glucose and lipid metabolism in several metabolic tissues, including adipose tissue and the liver. In hepatocytes, insulin regulates glucose uptake, promotes glycogenesis, and activates key regulators of de novo lipogenesis (DNL) while it simultaneously decreases gluconeogenesis, promoting glycogen storage [31][53]. On the other hand, in adipocytes, insulin has three main actions: (1) to promote the esterification of fatty acids, (2) to promote the storage of esterified fatty acids, including triglycerides (TGs), in lipid droplets, and (3) to inhibit lipolysis via hormone-sensitive lipase inactivation [32][54].
In an obese state and in NAFLD patients, systemic insulin resistance results in increased lipolysis and, thus, excess free fatty acids (FFAs), and inflammatory cytokines from peripheral adipose tissue can enter the liver through the portal circulation. The accumulation of FFAs and lipid metabolites in hepatocytes may induce a disruption of the insulin-signaling pathway and, subsequently, hepatic insulin resistance. Moreover, hepatic insulin resistance contributes to hyperglycemia, hyperinsulinemia, and increased lipid accumulation through DNL stimulation and mitochondrial fatty acid β-oxidation (FAO) inhibition, thus aggravating hepatic steatosis [33][34][55,56].
2.4. Progression of Nonalcoholic AFatty Liver Disease to Nonalcoholic SteatohepatitisLD to NASH
2.4.1. Lipotoxicity and Oxidative Stress
Several studies have highlighted that lipotoxicity leads to hepatocyte injury and the progression of NASH. The imbalance between lipid acquisition (increased uptake of circulating FFAs and DNL) and lipid exportation (downregulated FAO and export of lipids in very low density lipoproteins (VLDL)) further promotes lipid accumulation in the liver and the progression of hepatic steatosis [34][35][36][56,57,58]. In addition to FFAs, other types of lipids and their derivatives, including free cholesterol and ceramides, are involved in the development of liver lipotoxicity in NAFLD and NASH patients [37][38][59,60]. Lipotoxicity-induced hepatic injury leads to hepatocyte ballooning degeneration (liver cell swelling), fibrosis, and glomerular inflammation, which are considered to be the key histological features for NASH diagnosis [39][40][61,62].
In NAFLD patients, mitochondrial dysfunction plays a pivotal role during the transition from simple steatosis to NASH [41][63]. The energy homeostasis in hepatic cells is regulated by mitochondrial FAO, electron transfer, the production of ATP, and reactive oxygen species (ROS) [42][64]. Mitochondrial dysfunction contributes to an imbalance between prooxidant and antioxidant mechanisms, thus leading to lipid accumulation and excess ROS generation. The latter leads to the activation of inflammatory mediators and signaling pathways exacerbating inflammation, ROS generation, and oxidative DNA damage in NASH patients [43][44][65,66]. The overload of FFAs leads to an increase in the permeability of the inner mitochondrial membrane, 31–40% lower maximal respiration associated with mitochondrial uncoupling, electron leakage, augmented hepatic oxidative stress, and oxidative DNA damage [43][45][4,65].
2.4.2. Hepatic Inflammation and Fibrosis
The mechanistic concepts of inflammation in NAFLD/NASH have recently been reviewed and are associated with the exacerbated production of inflammatory factors from extrahepatic tissues (adipose tissue and gut) and in the liver by injured hepatocytes and the activation of resident hepatic macrophages, Kupffer cells [46][67]. Excess levels of circulating and hepatic FFA accumulation, altered gut microbiome, gut permeability alterations, the release of endotoxin, and adipose tissue dysfunction lead to hepatocyte injury and apoptosis, ROS generation, and inflammatory response, representing the initial steps of progression to NASH. Immunogenic stimuli, including damage-associated molecular patterns (DAMPs) released by injured hepatocytes and pathogen-associated molecular patterns (PAMPs), are recognized by the innate immune system through pattern recognition receptors (PRRs), such as toll-like receptors (TLRs) and NOD-like receptors (NLRs), and play a key role in NASH [47][68]. Being widely expressed in hepatic cells, activated TLRs, particularly TLR-4, recruit Kupffer cells, which release pro-inflammatory cytokines, including TNF-a, IL-1, IL-6, fibrogenic factors such as TGF-β, and the further activation of pro-inflammatory transcription factors (NF-κB) [48][69]. Cytokine signaling promotes the recruitment of immune effector cells, including neutrophils, dendritic cells, natural killer (NK) cells, and cytotoxic T cells, with subsequent hepatocyte injury via oxidative-stress-mediated mechanisms, indicating that adaptive immunity plays an important role in the progression of this disease [49][70]. In NASH patients, NOD-like receptor protein 3 (NLRP3) inflammasome is upregulated in injured hepatocytes, Kupffer cells, and liver sinusoidal endothelial cells and can be activated by DAMPs and PAMPs [50][51][71,72].
Chronic inflammation is associated with fibrosis, which can further progress to bridging fibrosis and cirrhosis. In response to liver injury, activated Kupffer cells, infiltrating monocytes, activated and aggregated platelets, and damaged hepatocytes release platelet-derived growth factor (PDGF) and TGF-β1, leading to HSC activation. Upon their activation, HSCs express NLRP3 inflammasome and transdifferentiate into fibroblasts or myofibroblast-like cells with proliferative, inflammatory, and migratory properties [52][73]. Upon HSC proliferation, components of the extracellular matrix are produced, including collagen type I and type III as well as tissue inhibitor of metalloproteinases 1 (TIMP-1), all contributing to fibrogenesis [53][54][74,75].
3. Association between NASH and Atherosclerosis: Inflammation and Oxidative Stress as Key Players
A large body of epidemiological and clinical evidence demonstrates that NAFLD is not only associated with liver morbidity but also with CVD development, arrhythmias, left ventricular dysfunction, and heart failure [55][56][57][76,77,78]. NAFLD and CVD are both manifestations of end-organ damage of MetS and share several common environmental and genetic factors [58][59][60][79,80,81] It has been demonstrated that NAFLD progression to NASH is associated with more severe atherosclerosis and is even considered an independent risk factor for coronary artery disease (CAD) [30][61][8,82]. Likewise, atherosclerosis accompanied by NAFLD/NASH has more adverse metabolic burdens than atherosclerosis alone. In this aspect, several studies provide evidence of a strong association between NASH and: (1) carotid atherosclerosis [62][63][64][65][83,84,85,86] and (2) subclinical manifestations of atherosclerosis in patients with or without T2DM, including increased intima–media thickness, endothelial dysfunction, arterial stiffness, impaired left ventricular function, reduced flow-mediated vasodilation, and coronary calcification [65][66][67][86,87,88]. These associations are independent of traditional cardiovascular risk factors and MetS characteristics across a wide range of patient populations [68][89]. Atherosclerosis is a progressive multifactorial disease characterized by the thickening of the arteries and endothelial dysfunction and is a main cause of myocardial infarction and stroke [69][70][90,91] Proinflammatory activation of endothelial cells (ECs) initiate the penetration of monocytes into the intima media, where they predominantly mature to pro-inflammatory macrophages (the M1 phenotype) that actively take up modified low-density lipoproteins (oxLDL) via scavenger receptors (e.g., scavenger receptor A1 (SR-A1), lectin-like oxLDL receptor-1 (LOX-1), and CD36) and release a variety of inflammatory cytokines and chemokines that are essential for the propagation of inflammation [71][92]. The excessive influx of modified LDLs and the accumulation of cholesterol esters in intimal macrophages due to the increased vascular permeability of the endothelial barrier lead to the generation of foam cells, which play a key role at all stages of atherosclerotic lesion development, from initial vascular lesions to advanced plaques [70][91]. Another main imbalance observed in atherosclerosis is the upregulation of acetyl-CoA acetyltransferase (ACAT1), the enzyme responsible for cholesterol esterification, and the downregulation of neutral cholesterol ester hydrolase (NCEH), the enzyme responsible for the hydrolysis of cholesterol esters to free cholesterol, resulting in the accumulation of cholesterol esters and the further transformation of macrophages to foam cells. Yellow foam cells aggregate on the arterial walls and cause the development of fatty streaks, which form a fibrous atherosclerotic plaque cap [72][73][93,94]. At advanced stages of the disease, growth factor released by macrophages in the plaque causes the proliferation of smooth muscle cells and the plaque becomes fibrotic [74][95]. Activated macrophages and T lymphocytes of the fibrous atherosclerotic plaque cap stimulate the production of proteolytic metalloproteases, leading to the degradation of the extracellular matrix by phagocytosis and to a decrease in the stability of the fibrous cap. Plaque rupture leads to a coagulation process, blood clot formation, thrombus formation, and a blockade of the arteries [75][96]. These atherogenic processes are triggered by well-identified risk factors, such as hypertension, hyperlipidemia, and diabetes mellitus. NAFLD can contribute to and aggravate atherosclerosis development, but the precise mechanisms remain unclear. The supposed mechanisms for accelerating atherosclerotic disease in patients with NASH are very complex and include, among others, chronic inflammation, lipid accumulation, and oxidative stress (Figure 1). The following sections display possible linkages between these conditions at the molecular level.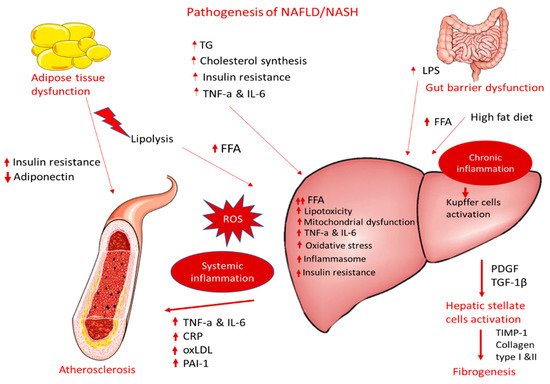
Figure 1.
Pathogenesis of NAFLD/NASH and association with atherosclerosis.
3.1. Dyslipidemia and Lipotoxicity
As mentioned before, NAFLD/NASH is characterized by hepatic fat accumulation, which results from an imbalance between lipid acquisition and lipid disposal that is mediated by increased hepatic DNL and the uptake of circulating FFAs, a downregulation of compensatory FAO, and an altered export of lipids in VLDL. This imbalance is considered to be the initiating mechanism of atherosclerosis as well [76][97]. Moreover, patients with NAFLD exert typical atherogenic dyslipidemia features, including higher serum TG and oxLDL levels and lower serum HDL. Moreover, high serum oxLDL levels are more dominant in patients with atherosclerosis due to their localization in macrophage-derived foam cells. It has been reported that TNF-a is implicated in the decrease in HDL levels, indicating a link between inflammation and the development and progression of insulin-resistant conditions, including NASH and atherosclerosis [77][78][98,99].3.2. Adipose Tissue Dysfunction
Adipose tissue increases CVD risk by inducing many obesity-associated complications, such as dyslipidemia, high blood pressure, insulin resistance, and T2DM [79][100]. Visceral adiposity causes an increase in the hepatic accumulation of FFAs, accompanied by decreased FFA oxidation and altered glucose metabolism, contributing to hepatic insulin resistance. Visceral obesity is associated with proinflammatory cytokine production, i.e., TNF-α, IL-6, monocyte chemoattractant protein-1 (MCP-1), CRP, adipokines, and macrophage infiltration, resulting in local and systemic inflammation. The latter is associated with the consequent hepatic production of pro-atherogenic molecules, such as plasminogen activator inhibitor-1 (PAI-1) and fibrinogen, thereby causing endothelial dysfunction and increasing the risk of atherothrombosis. A consistent and ever-growing line of research has revealed a link between intestinal dysbiosis, inflamed adipose tissue, and NAFLD with atherosclerosis and other cardiac complications. The progression of NAFLD to NASH leads to the production of proinflammatory cytokines, atherogenic lipoproteins, and vasoactive and thrombogenic factors and enhanced oxidative stress, resulting in an increased risk of atherosclerosis and myocardial infarction.3.3. Endothelial Dysfunction and Inflammation
The endothelial dysfunction and abnormal vasoreactivity observed in NASH patients lead to chronic inflammation, increased vasoconstriction, and increased prothrombotic factor production, thus elevating the risk of atherosclerosis and several other cardiovascular implications [80][101]. Inflammation plays a central role in both NASH and atherosclerosis, involving the local presence or resident macrophages. Macrophages accumulate oxidized lipoproteins through the SRs and lead to the generation of foam cells and the release of cytokines during atherogenesis. Similarly, in NASH the resident hepatic macrophages, Kupffer cells, take up modified lipoproteins expressing SR CD36 and, thus, further contribute to atherosclerotic lesions [72][78][93,99]. Specifically, immunogenic stimuli, including the accumulation of modified triglyceride-rich lipoproteins, serve as DAMPs and activate TLRs recruiting macrophages and resident Kupffer cells. The activated NLRP3 inflammasome leads to the release of pro-inflammatory cytokines, including TNF-a, IL-1, and IL-6; fibrogenic factors, such as TGF-β; and the further activation of pro-inflammatory transcription factors (NF-κB) [48][69], providing an important link between NASH, liver fibrosis, and the development of vascular damage and atherosclerosis [81][102].4. Explaining the Effectiveness of Incretin-Based Drugs on NASH and Atherosclerosis
4.1. Potential Mechanisms of GLP-1 RAs’ Effectiveness in NASH
The only GLP-1 RAs that have been proven to resolve NASH based on histological data are liraglutide and semaglutide. These GLP-1 RAs, apart from their glucose-lowering effect, have demonstrated an ability to significantly reduce body weight, with semaglutide having a more pronounced effect. This effect is mainly attributed to a modulation of appetite and a feeling of satiety as well as reduced caloric intake through actions in the central nervous system combined with a reduction in glucosuria due to enhanced glycemic control (Figure 2) [82][145]. Most of the weight that is being lost during treatment with GLP-1 RAs is fat mass, particularly visceral fat, due to their effect on adipose tissue [83][146]. However, although body weight reduction is a key parameter in NASH resolution, it cannot solely explain the improved liver function observed in patients treated with GLP-1 RAs. Specifically, in a study performed by Shiomi et al. (2020) in Japanese patients with T2DM and NAFLD receiving liraglutide for 24 weeks the improvement in liver function or fibrosis (assessed through aspartate aminotransferase, alanine aminotransferase, and fibrosis-4 indices) was found to be independent of the body mass index [84][147]. Several other mechanisms are involved, as demonstrated in Figure 2.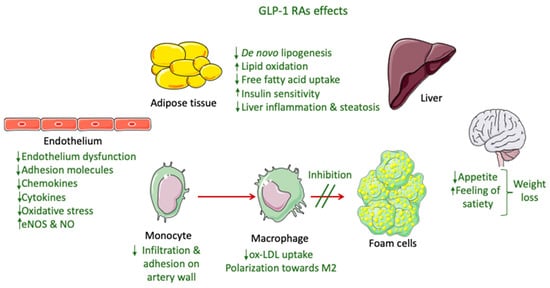
Figure 2.
GLP-1 RA effects involved in NASH and atherosclerosis improvement.