Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 3 by Conner Chen and Version 2 by Conner Chen.
Pentameric ligand-gated ion channels (pLGICs) mediate or modulate fast synaptic communication in the central and peripheral nervous systems making them vital for neurological processes ranging from memory and learning to nicotine addiction.
- pentameric ligand-gated ion channels
- lipid binding sites
- l
1. Introduction
Pentameric ligand-gated ion channels (pLGICs) mediate or modulate fast synaptic communication in the central and peripheral nervous systems making them vital for neurological processes ranging from memory and learning to nicotine addiction [1][2][3][4]. pLGICs respond to the binding of neurotransmitters by transiently opening either cation- or anion-selective ion channels across the post-synaptic membrane, with prolonged exposure favoring a non-conductive desensitized state(s). The relative stabilities of the resting, open and desensitized states, as well as the rates of inter-conversation between them, shape the magnitude and temporal nature of the agonist-induced response to establish effective inter-neuronal or neuromuscular communication. pLGICs are also targeted by a variety of exogenous molecules that allosterically modulate the agonist-induced response in a manner that alters synaptic communication [5][6].
Lipids are potent activators and/or modulators of ion channels including inward rectifying potassium channels, voltage-gated potassium channels, transient receptor potential channels, mechanosensitive ion channels and pLGICs [7]. The functional sensitivity of pLGICs to membrane lipids was first shown in the 1970s through studies that sought to identify the structural features in the muscle-type Torpedo nicotinic acetylcholine receptor (nAChR) that are responsible for both agonist binding and channel gating. These studies showed that to retain both binding and gating, cholate-solubilized receptors must be purified in the presence of lipids and then placed in a bilayer with a particular lipid composition [8][9]. Since then, the effects of lipids on Torpedo nAChR function have been characterized extensively [10][11][12]. More recently, studies of pLGIC–lipid interactions have extended to other members of the super-family, including the prokaryotic Gleobacter violaceus ligand-gated ion channel, GLIC, and Erwinia chrysanthemi (now Dickeya dadantii) ligand gated ion channel, ELIC.
Over the past 15 years, increasing numbers of structures have shed light on the modes of lipid binding to pLGICs, thus providing a structural context for interpreting functional data on pLGIC–lipid interactions [13].
2. pLGIC Structure
Both eukaryotic and prokaryotic pLGICs exhibit a common architecture consisting of five subunits arranged either symmetrically (homomeric) or pseudo-symmetrically (heteromeric) around a central ion channel pore (Figure 1). In humans, there are four main families of pLGICs that conduct either cations or anions leading to either excitatory or inhibitory post-synaptic responses, respectively. The excitatory cation-selective pLGICs respond to the neurotransmitters, acetylcholine (nicotinic acetylcholine receptor, nAChR) and serotonin (serotonin receptor, 5-HT3R), while the inhibitory anion-selective pLGICs respond to γ-aminobutyric acid (GABA receptor, GABAAR) and glycine (glycine receptor, GlyR). Humans also uniquely express a cation-selective zinc-activated channel, ZAC [14]. Each of the four main families includes a variety of functional hetero- and homo-pentamers that form from different combinations of the sixteen distinct nAChR subunits (α1–α7, α9–α10, β1–β4, δ, γ, and ε), the five distinct 5-HT3AR subunits (A–E), the nineteen distinct GABAAR subunits (α1–α6, β1–β3,γ1–γ3, ε, δ, π, θ, and ρ1–ρ3) or the five distinct GlyR subunits (α1–α4, and β). Each combination leads to a pLGIC with a unique electrophysiological and pharmacological fingerprint. Receptors with different subunit are also targeted to specific cell types and/or regions of the brain [15].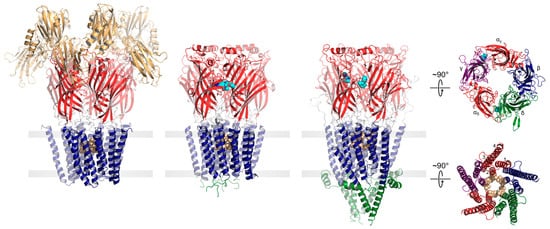
Figure 1. pLGICs display a conserved core architecture with diverse auxiliary features. Side views of the prokaryote DeCLIC (PDB: 6V4S, far left), the human α1β3γ2 GABAAR (PDB: 7QNE, middle left), and the Torpedo nAChR (PDB: 7QL5, middle right) colored according to domains (NTD, orange; ECD, red; TMD, blue; ICD, green). Bound agonists are presented as cyan spheres at the interfaces between two subunits. In the side views, the principal subunit is on the left and the complementary subunit is on the right. Residues forming the channel gate are presented as tan spheres. Top-down views of the Torpedo nAChR ECD (top) and TMD (bottom) are shown on the far right colored according to subunit (α, red; β, blue; γ, purple; δ, green).
3. Nicotinic Acetylcholine Receptors
3.1. Functional Sensitivity of the nAChR to Lipids
Cell-based assays and mutagenesis indirectly suggest that numerous members of the nAChR family exhibit a functional sensitivity to lipids [33][34][35][36][37][38][39][40]. In addition, functional measurements using the Torpedo nAChR reconstituted into liposomes with defined lipid compositions show definitively that a broad range of lipids influence the agonist-induced response, and do so through complex mechanisms. For example, ternary lipid mixtures containing phosphatidylcholine (PC) and both cholesterol and anionic lipids support a robust agonist-induced response, while PC membranes lacking both lipids lock the nAChR in a non-responsive uncoupled conformation that binds agonist but does not normally undergo agonist-induced conformational transitions [41]. The above observation was interpreted to suggest that both cholesterol and anionic lipids are essential for nAChR function and that both lipids exert their functional effects by binding to distinct allosteric sites, a view still prevalent in the literature. Three subsequent observations, however, suggested that neither lipid/lipid-type is essential for the nAChR to undergo agonist-induced conformational transitions. First, increasing levels of either cholesterol or the anionic lipid, phosphatidic acid (PA), in a PC membrane stabilize an increasing proportion of agonist-responsive nAChRs, although PA is more effective in this regard [42][43]. Second, in the presence of anionic lipids a variety of neutral lipids substitute for cholesterol in supporting nAChR function [13][44][45]. Finally, in the presence of cholesterol a variety of anionic lipids substitute for PA in supporting a functional nAChR. Collectively these observations show that if both cholesterol and anionic lipids influence function by binding to distinct allosteric sites, then the lipid specificities for these sites are low and their occupancies not absolutely required for an agonist-induced response. There are also intriguing differences in the capacities of anionic lipids to influence the agonist-induced response. For example, PC membranes containing high levels of PA stabilize a large proportion of agonist-responsive nAChRs, while PC membranes containing similar levels of phosphatidylserine (PS) or other anionic lipids do not [46][47][48]. These and other observations [43] suggest that PA has a unique capacity to stabilize an agonist responsive nAChR. One possibility is that the small anionic headgroup allows PA to bind with higher affinity and thus greater occupancy to an allosteric site to promote channel function. Another is that high levels of PA increase the ordering of the surrounding bilayer, possibly in a manner that mimics the ordering observed in the presence of cholesterol [48]. High levels (40 mol%) of PA in a PC membrane may be particularly effective at stabilizing a functional nAChR because PA exhibits both the required anionic headgroup charge and an ability to influence bulk membrane physical properties in a manner that supports agonist-induced conformational transitions. Further supporting a role for bulk membrane physical properties in nAChR function, hydrophobically thick PC membranes promote conformational transitions even in the absence of cholesterol and anionic lipids [49].3.2. Sites of Lipid Action at the nAChR
Early biophysical and computational studies suggested that cholesterol, and possibly other lipids, bind to both annular and non-annular sites on the nAChR to influence function [50][51][52]. Annular sites are those located at the periphery of the TMD in rapid exchange with lipids in the bulk membrane environment, while non-annular sites are those buried between TMD α-helices that are shielded from bulk membrane lipids [53]. In contrast to the plethora of annular lipid sites observed in nAChR structures (discussed below), none of the nAChR structures solved to date exhibits density attributed to buried non-annular lipids. Both the abundance of observed annular lipid sites, which should be more mobile than non-annular lipids, and the absence of observed buried non-annular lipids argue against the existence of functional non-annular lipid binding to the nAChR. On the other hand, structures of the nAChR and other pLGICs reveal density due to annular lipids, but with the acyl chains extending in between TMD 𝛼-helices (see below). In these cases, the distinction between annular and non-annular lipid binding is blurred. The first direct structural evidence for annular lipid binding to the nAChR was obtained from cryo-electron microscopy (cryo-EM) structures of the detergent-solubilized human neuronal 𝛼4𝛽2 nAChR (both 𝛼43𝛽22 and 𝛼42𝛽23 stoichiometries) and the azolectin nanodisc-reconstituted human neuronal 𝛼3𝛽4 nAChR, the two sets of structures solved in the presence of the water-soluble cholesterol analog, cholesterol hemisuccinate (CHS) [54][55]. Each structure exhibits regions of electron density at the periphery of the TMD that was modeled as cholesterol (Figure 2). The bound cholesterol, located in the cytoplasmic leaflet at both the M4–M1 and the M4–M3 interfaces of each subunit, is close to residues covalently labeled in the Torpedo nAChR by a photoactivatable cholesterol probe [56]. Notably, the electron density attributed to cholesterol disappears when the cryo-EM samples are prepared in the absence of CHS.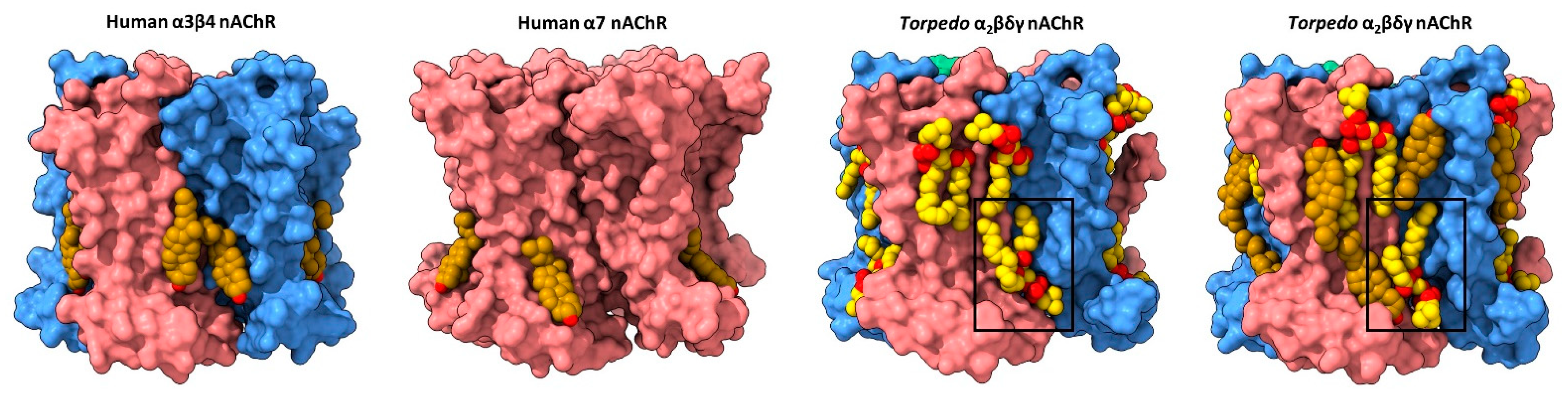
Figure 2. Lipid binding to both extracellular and intracellular leaflet sites on the nAChR. Side views of the TMD for the cholesterol-bound α3β4 nAChR (PDB: 6PV7, far left) and α7 nAChR (PDB: 7EKI, middle left), phospholipid-bound Torpedo nAChR (PDB: 7QL5, middle right), and cholesterol- and phospholipid-bound Torpedo nAChR (PDB: 7SMQ, far right) represented as surfaces, with principal and complementary subunits colored in pink and blue, respectively. Bound cholesterol (brown) and phospholipids (yellow) are presented as spheres with oxygen, nitrogen, and phosphorus colored in red, blue, and orange, respectively.
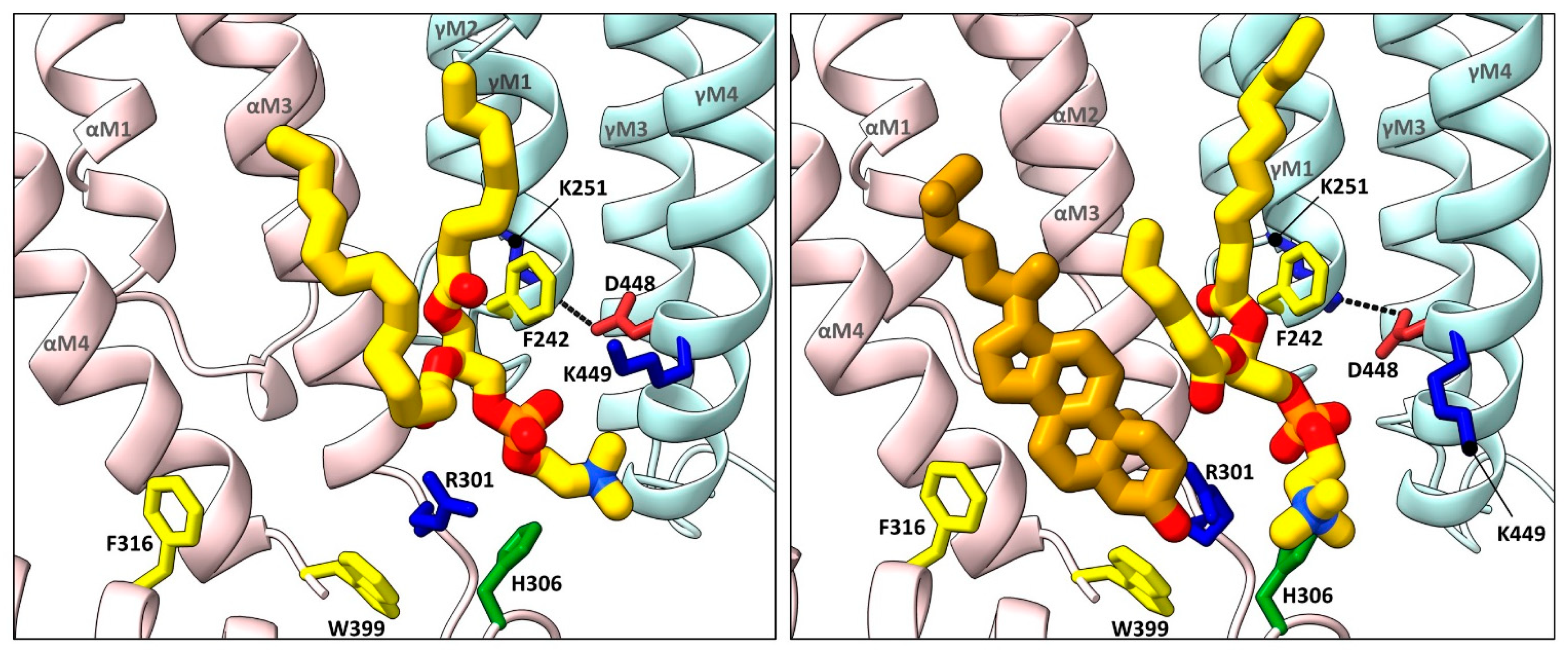
Figure 3. Cholesterol and phospholipids bind to adjacent or overlapping sites in Torpedo nAChR structures. Zoomed in views (defined in Figure 2) of Torpedo nAChR structures with bound phospholipids (PDB: 7QL5) or both phospholipids and cholesterol (PDB: 7SMQ). Subunits and lipids are colored as in Figure 2, with residues interacting with bound lipids represented as sticks colored according to residue type (non-polar, tan; aromatic, yellow; polar, green; cationic, blue; anionic, red). The M2–M4 salt bridge adjacent to the bound lipids is shown as a dashed line.
References
- Rodríguez Cruz, P.M.; Cossins, J.; Beeson, D.; Vincent, A. The neuromuscular junction in health and disease: Molecular mechanisms governing synaptic formation and homeostasis. Front. Mol. Neurosci. 2020, 13, 610964.
- Gibbs, E.; Chakrapani, S. Structure, function and physiology of 5-hydroxytryptamine receptors subtype 3. In Macromolecular Protein Complexes III: Structure and Function. Subcellular Biochemistry; Harris, J.R., Marles-Wright, J., Eds.; Springer International Publishing: Cham, Switzerland, 2021; pp. 373–408. ISBN 9783030589714.
- Lara, C.O.; Burgos, C.F.; Moraga-Cid, G.; Carrasco, M.A.; Yévenes, G.E. Pentameric ligand-gated ion channels as pharmacological targets against chronic pain. Front. Pharmacol. 2020, 11, 167.
- Koukouli, F.; Changeux, J.-P. Do nicotinic receptors modulate high-order cognitive processing? Trends Neurosci. 2020, 43, 550–564.
- Zhang, Y.; Wang, K.; Yu, Z. Drug development in channelopathies: Allosteric modulation of ligand-gated and voltage-gated ion channels. J. Med. Chem. 2020, 63, 15258–15278.
- Cheng, W.W.L.; Arcario, M.J.; Petroff, J.T., II. Druggable lipid binding sites in pentameric ligand-gated ion channels and transient receptor potential channels. Front. Physiol. 2022, 12, 798102.
- Thompson, M.J.; Baenziger, J.E. Ion channels as lipid sensors: From structures to mechanisms. Nat. Chem. Biol. 2020, 16, 1331–1342.
- Fong, T.M.; McNamee, M.G. Correlation between acetylcholine receptor function and structural properties of membranes. Biochemistry 1986, 25, 830–840.
- Criado, M.; Eibl, H.; Barrantes, F.J. Effects of lipids on acetylcholine receptor. essential need of cholesterol for maintenance of agonist-induced state transitions in lipid vesicles. Biochemistry 1982, 21, 3622–3629.
- Baenziger, J.E.; Hénault, C.M.; Therien, J.P.D.; Sun, J. Nicotinic acetylcholine receptor–Lipid interactions: Mechanistic insight and biological function. Biochim. Biophys. Acta Biomembr. 2015, 1848, 1806–1817.
- Baenziger, J.E.; Domville, J.A.; Therien, J.P.D. The role of cholesterol in the activation of nicotinic acetylcholine receptors. In Current Topics in Membranes; Levitan, I., Ed.; Academic Press: New York, NY, USA, 2017; Volume 80, pp. 95–137. ISBN 9780128093887.
- Barrantes, F.J. Phylogenetic conservation of protein–Lipid motifs in pentameric ligand-gated ion channels. Biochim. Biophys. Acta Biomembr. 2015, 1848, 1796–1805.
- Thompson, M.J.; Baenziger, J.E. Structural basis for the modulation of pentameric ligand-gated ion channel function by lipids. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183304.
- Madjroh, N.; Mellou, E.; Æbelø, L.; Davies, P.A.; Söderhielm, P.C.; Jensen, A.A. Probing the molecular basis for signal transduction through the zinc-activated channel (ZAC). Biochem. Pharmacol. 2021, 193, 114781.
- Sparling, B.A.; DiMauro, E.F. Progress in the discovery of small molecule modulators of the cys-loop superfamily receptors. Bioorg. Med. Chem. Lett. 2017, 27, 3207–3218.
- Hassaine, G.; Deluz, C.; Grasso, L.; Wyss, R.; Tol, M.B.; Hovius, R.; Graff, A.; Stahlberg, H.; Tomizaki, T.; Desmyter, A.; et al. X-ray structure of the mouse serotonin 5-HT3 receptor. Nature 2014, 512, 276–281.
- Sente, A.; Desai, R.; Naydenova, K.; Malinauskas, T.; Jounaidi, Y.; Miehling, J.; Zhou, X.; Masiulis, S.; Hardwick, S.W.; Chirgadze, D.Y.; et al. Differential assembly diversifies GABA a receptor structures and signalling. Nature 2022, 604, 190–194.
- Zhu, H.; Gouaux, E. Architecture and assembly mechanism of native glycine receptors. Nature 2021, 599, 513–517.
- Hu, H.; Howard, R.J.; Bastolla, U.; Lindahl, E.; Delarue, M. Structural basis for allosteric transitions of a multidomain pentameric ligand-gated ion channel. Proc. Natl. Acad. Sci. USA 2020, 117, 13437–13446.
- Lee, W.-Y.; Sine, S.M. Principal pathway coupling agonist binding to channel gating in nicotinic receptors. Nature 2005, 438, 243–247.
- Gupta, S.; Chakraborty, S.; Vij, R.; Auerbach, A. A mechanism for acetylcholine receptor gating based on structure, coupling, phi, and flip. J. Gen. Physiol. 2017, 149, 85–103.
- Lee, W.-Y.; Free, C.R.; Sine, S.M. Nicotinic receptor interloop proline anchors Β1-Β2 and cys loops in coupling agonist binding to channel gating. J. Gen. Physiol. 2008, 132, 265–278.
- Beckstein, O.; Tai, K.; Sansom, M.S.P. Not ions alone: Barriers to ion permeation in nanopores and channels. J. Am. Chem. Soc. 2004, 126, 14694–14695.
- Nemecz, Á.; Prevost, M.S.; Menny, A.; Corringer, P.-J. Emerging molecular mechanisms of signal transduction in pentameric ligand-gated ion channels. Neuron 2016, 90, 452–470.
- Unwin, N.; Fujiyoshi, Y. Gating movement of acetylcholine receptor caught by plunge-freezing. J. Mol. Biol. 2012, 422, 617–634.
- Du, J.; Lü, W.; Wu, S.; Cheng, Y.; Gouaux, E. Glycine receptor mechanism elucidated by electron cryo-microscopy. Nature 2015, 526, 224–229.
- Polovinkin, L.; Hassaine, G.; Perot, J.; Neumann, E.; Jensen, A.A.; Lefebvre, S.N.; Corringer, P.-J.; Neyton, J.; Chipot, C.; Dehez, F.; et al. Conformational transitions of the serotonin 5-HT3 receptor. Nature 2018, 563, 275–279.
- Masiulis, S.; Desai, R.; Uchański, T.; Serna Martin, I.; Laverty, D.; Karia, D.; Malinauskas, T.; Zivanov, J.; Pardon, E.; Kotecha, A.; et al. GABAA receptor signalling mechanisms revealed by structural pharmacology. Nature 2019, 565, 454–459.
- Boyd, N.D.; Cohen, J.B. Kinetics of binding of acetylcholine to torpedo postsynaptic membranes: Association and dissociation rate constants by rapid mixing and ultrafiltration. Biochemistry 1980, 19, 5353–5358.
- Gielen, M.; Corringer, P.-J. The dual-gate model for pentameric ligand-gated ion channels activation and desensitization. J. Physiol. 2018, 596, 1873–1902.
- Bouzat, C.; Bartos, M.; Corradi, J.; Sine, S.M. The interface between extracellular and transmembrane domains of homomeric cys-loop receptors governs open-channel lifetime and rate of desensitization. J. Neurosci. 2008, 28, 7808–7819.
- Hénault, C.M.; Govaerts, C.; Spurny, R.; Brams, M.; Estrada-Mondragon, A.; Lynch, J.W.; Bertrand, D.; Pardon, E.; Evans, G.L.; Woods, K.; et al. A lipid site shapes the agonist response of a pentameric ligand-gated ion channel. Nat. Chem. Biol. 2019, 15, 1156–1164.
- Colón-Sáez, J.O.; Yakel, J.L. The A7 nicotinic acetylcholine receptor function in hippocampal neurons is regulated by the lipid composition of the plasma membrane. J. Physiol. 2011, 589, 3163–3174.
- Zhu, D.; Xiong, W.C.; Mei, L. Lipid rafts serve as a signaling platform for nicotinic acetylcholine receptor clustering. J. Neurosci. 2006, 26, 4841–4851.
- Báez-Pagán, C.A.; del Hoyo-Rivera, N.; Quesada, O.; Otero-Cruz, J.D.; Lasalde-Dominicci, J.A. Heterogeneous inhibition in macroscopic current responses of four nicotinic acetylcholine receptor subtypes by cholesterol enrichment. J. Membr. Biol. 2016, 249, 539–549.
- Santiago, J.; Guzmán, G.R.; Rojas, L.V.; Marti, R.; Asmar-Rovira, G.A.; Santana, L.F.; McNamee, M.G.; Lasalde-Dominicci, J.A. Probing the effects of membrane cholesterol in the torpedo californica acetylcholine receptor and the novel lipid-exposed mutation αc418w in xenopus oocytes. J. Biol. Chem. 2001, 276, 46523–46532.
- Li, L.; Lee, Y.H.; Pappone, P.; Palma, A.; McNamee, M.G. Site-specific mutations of nicotinic acetylcholine receptor at the lipid-protein interface dramatically alter ion channel gating. Biophys. J. 1992, 62, 61–63.
- Guzmán, G.R.; Ortiz-Acevedo, A.; Ricardo, A.; Rojas, L.V.; Lasalde-Dominicci, J.A. The polarity of lipid-exposed residues contributes to the functional differences between torpedo and muscle-type nicotinic receptors. J. Membr. Biol. 2006, 214, 131–138.
- da Costa Couto, A.R.G.M.; Price, K.L.; Mesoy, S.; Capes, E.; Lummis, S.C.R. The M4 helix is involved in α7 nach receptor function. ACS Chem. Neurosci. 2020, 11, 1406–1412.
- Mesoy, S.M.; Lummis, S.C.R. M4, the outermost helix, is extensively involved in opening of the A4β2 NACh receptor. ACS Chem. Neurosci. 2021, 12, 133–139.
- daCosta, C.J.B.; Baenziger, J.E. A Lipid-Dependent Uncoupled Conformation of the Acetylcholine Receptor. J. Biol. Chem. 2009, 284, 17819–17825.
- Baenziger, J.E.; Morris, M.L.; Darsaut, T.E.; Ryan, S.E. Effect of membrane lipid composition on the conformational equilibria of the nicotinic acetylcholine receptor. J. Biol. Chem. 2000, 275, 777–784.
- Hamouda, A.K.; Sanghvi, M.; Sauls, D.; Machu, T.K.; Blanton, M.P. Assessing the lipid requirements of the torpedo californica nicotinic acetylcholine receptor. Biochemistry 2006, 45, 4327–4337.
- Sunshine, C.; McNamee, M.G. Lipid modulation of nicotinic acetylcholine receptor function: The role of neutral and negatively charged lipids. BBA Biomembr. 1992, 1108, 240–246.
- Addona, G.H.; Sandermann, H.; Kloczewiak, M.A.; Miller, K.W. Low chemical specificity of the nicotinic acetylcholine receptor sterol activation site. Biochim. Biophys. Acta Biomembr. 2003, 1609, 177–182.
- daCosta, C.J.B.; Medaglia, S.A.; Lavigne, N.; Wang, S.; Carswell, C.L.; Baenziger, J.E. Anionic lipids allosterically modulate multiple nicotinic acetylcholine receptor conformational equilibria. J. Biol. Chem. 2009, 284, 33841–33849.
- daCosta, C.J.B.; Wagg, I.D.; McKay, M.E.; Baenziger, J.E. Phosphatidic acid and phosphatidylserine have distinct structural and functional interactions with the nicotinic acetylcholine receptor. J. Biol. Chem. 2004, 279, 14967–14974.
- daCosta, C.J.B.; Ogrel, A.A.; McCardy, E.A.; Blanton, M.P.; Baenziger, J.E. Lipid-protein interactions at the nicotinic acetylcholine receptor. A functional coupling between nicotinic receptors and phosphatidic acid-containing lipid bilayers. J. Biol. Chem. 2002, 277, 201–208.
- daCosta, C.J.B.; Dey, L.; Therien, J.P.D.; Baenziger, J.E. A distinct mechanism for activating uncoupled nicotinic acetylcholine receptors. Nat. Chem. Biol. 2013, 9, 701–707.
- Jones, O.T.; McNamee, M.G. Annular and nonannular binding sites for cholesterol associated with the nicotinic acetylcholine receptor. Biochemistry 1988, 27, 2364–2374.
- Antollini, S.S.; Barrantes, F.J. Disclosure of discrete sites for phospholipid and sterols at the protein—Lipid interface in native acetylcholine receptor-rich membrane. Biochemistry 1998, 37, 16653–16662.
- Brannigan, G.; Hénin, J.; Law, R.; Eckenhoff, R.G.; Klein, M.L. Embedded cholesterol in the nicotinic acetylcholine receptor. Proc. Natl. Acad. Sci. USA 2008, 105, 14418–14423.
- Lee, A.G. How lipids affect the activities of integral membrane proteins. Biochim. Biophys. Acta Biomembr. 2004, 1666, 62–87.
- Walsh, R.M.; Roh, S.-H.; Gharpure, A.; Morales-Perez, C.L.; Teng, J.; Hibbs, R.E. Structural principles of distinct assemblies of the human A4β2 nicotinic receptor. Nature 2018, 557, 261–265.
- Gharpure, A.; Teng, J.; Zhuang, Y.; Noviello, C.M.; Walsh, R.M.; Cabuco, R.; Howard, R.J.; Zaveri, N.T.; Lindahl, E.; Hibbs, R.E. Agonist selectivity and ion permeation in the A3β4 ganglionic nicotinic receptor. Neuron 2019, 104, 501–511.
- Hamouda, A.K.; Chiara, D.C.; Sauls, D.; Cohen, J.B.; Blanton, M.P. Cholesterol interacts with transmembrane α-helices M1, M3, and M4 of the torpedo nicotinic acetylcholine receptor: Photolabeling studies using azichoiesterol. Biochemistry 2006, 45, 976–986.
- Rahman, M.; Basta, T.; Teng, J.; Lee, M.; Worrell, B.T.; Stowell, M.H.B.; Hibbs, R.E. Structural mechanism of muscle nicotinic receptor desensitization and block by curare. Nat. Struct. Mol. Biol. 2022, 29, 386–394.
- Unwin, N. Segregation of lipids near acetylcholine-receptor channels imaged by cryo-EM. IUCrJ 2017, 4, 393–399.
- Unwin, N. Protein–lipid architecture of a cholinergic postsynaptic membrane. IUCrJ 2020, 7, 852–859.
- Unwin, N. Protein–lipid interplay at the neuromuscular junction. Microscopy 2022, 71, i66–i71.
- Rahman, M.; Teng, J.; Worrell, B.T.; Karlin, A.; Stowell, M.H.B.; Hibbs, R.E.; Noviello, C.M.; Lee, M. Structure of the native muscle-type nicotinic receptor and inhibition by snake venom toxins article structure of the native muscle-type nicotinic receptor and inhibition by snake venom toxins. Neuron 2020, 106, 952–962.
- Zarkadas, E.; Pebay-Peyroula, E.; Thompson, M.J.; Schoehn, G.; Uchański, T.; Steyaert, J.; Chipot, C.; Dehez, F.; Baenziger, J.E.; Nury, H. Conformational transitions and ligand-binding to a muscle-type acetylcholine receptor. Neuron 2022, 110, 1358–1370.
- Strikwerda, J.R.; Sine, S.M. Unmasking coupling between channel gating and ion permeation in the muscle nicotinic receptor. Elife 2021, 10, e66225.
- Noviello, C.M.; Gharpure, A.; Mukhtasimova, N.; Borek, D.; Sine, S.M.; Hibbs, R.E.; Cabuco, R.; Baxter, L. Structure and gating mechanism of the a7 nicotinic acetylcholine receptor. Cell 2021, 184, 2121–2134.
- Zhao, Y.; Liu, S.; Zhou, Y.; Zhang, M.; Chen, H.; Eric Xu, H.; Sun, D.; Liu, L.; Tian, C. Structural basis of human A7 nicotinic acetylcholine receptor activation. Cell Res. 2021, 31, 713–716.
- Flayhan, A.; Mertens, H.D.T.; Ural-Blimke, Y.; Martinez Molledo, M.; Svergun, D.I.; Löw, C. Saposin lipid nanoparticles: A highly versatile and modular tool for membrane protein research. Structure 2018, 26, 345–355.
- Reddy, B.; Bavi, N.; Lu, A.; Park, Y.; Perozo, E. Molecular basis of force-from-lipids gating in the mechanosensitive channel MscS. Elife 2019, 8, e50486.
More