Fts-Z is being explored as potent target for inhibiting emerging microbes due to its central role in Z-ring formation and conserved nature in nearly all bacterial species and absence in higher eukaryotes. Its absence in bacteria induces filamentation and cell death occurs. Antimicrobial resistance (AMR) is a pressing issue worldwide that must be addressed swiftly. It is driven by spontaneous evolution, bacterial mutation, and the dissemination of resistant genes via horizontal gene transfer. Researchers are working on many novel targets, which can become a pathway to inhibit harmful bacteria. Filamentous Thermosensitive mutant-Z (Fts-Z) is one such bacterial target that has gained popularity amongst scientists due to its conserved nature in bacteria and absence in eukaryotes.
- antimicrobials
- cytokinesis
- Fts-Z
1. Berberine and Derivatives
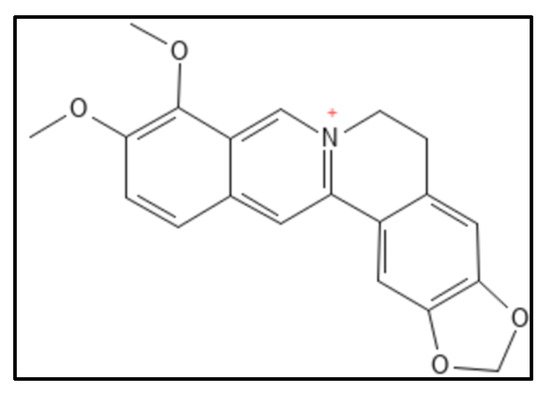
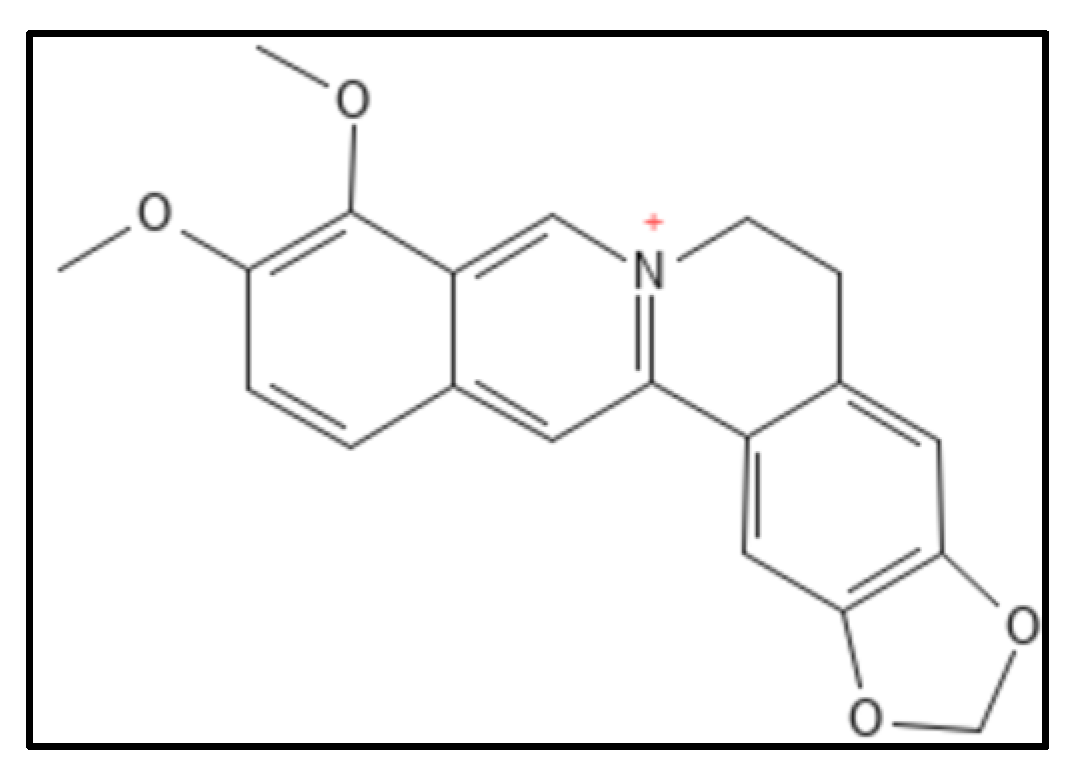
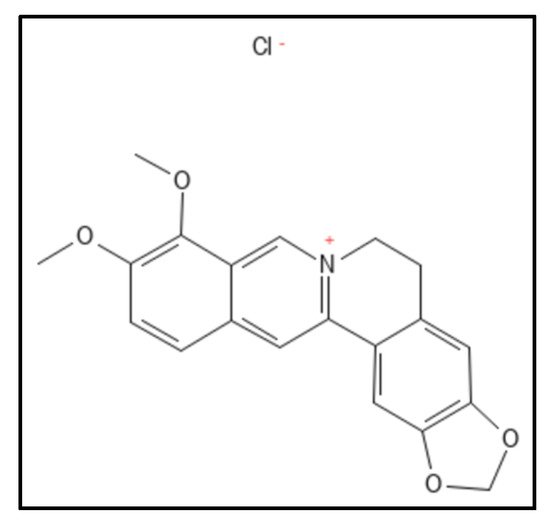
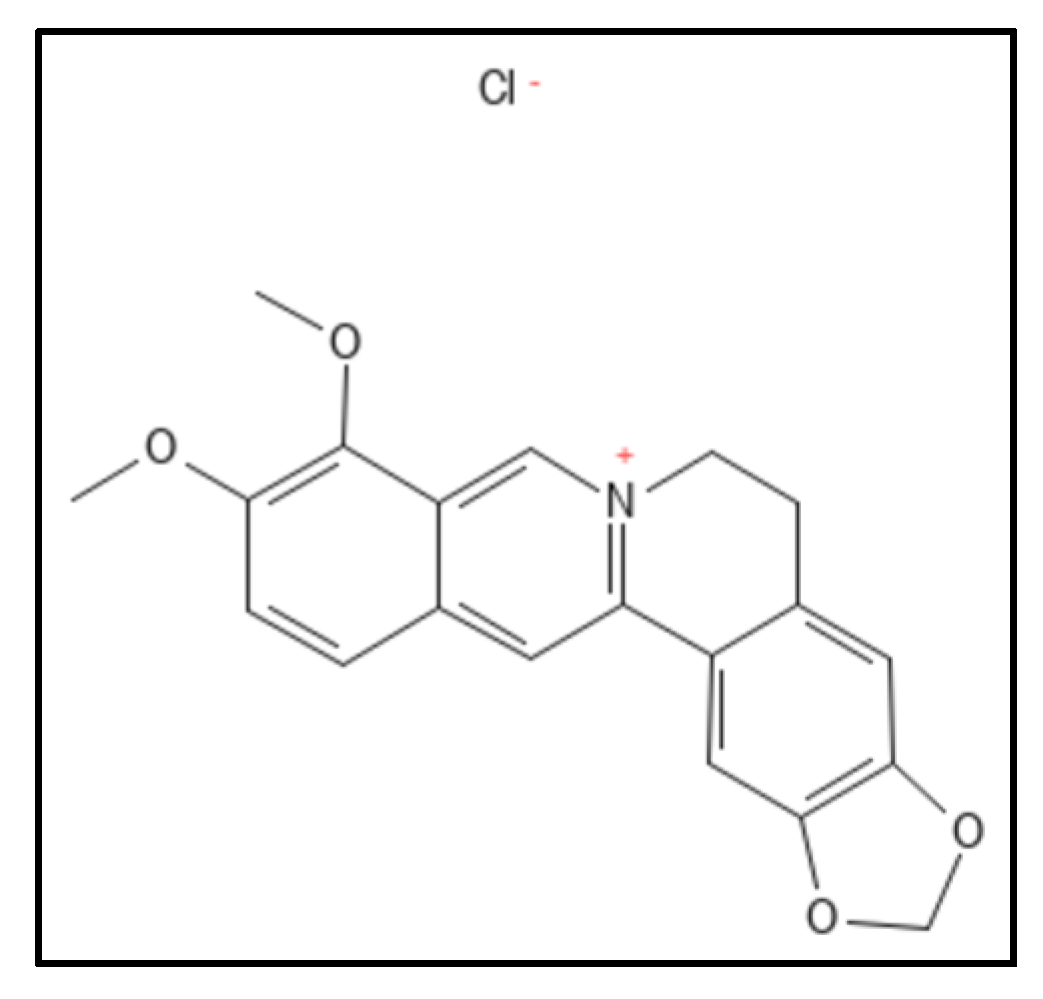
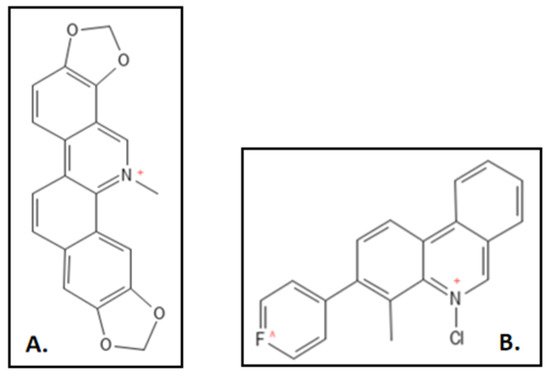
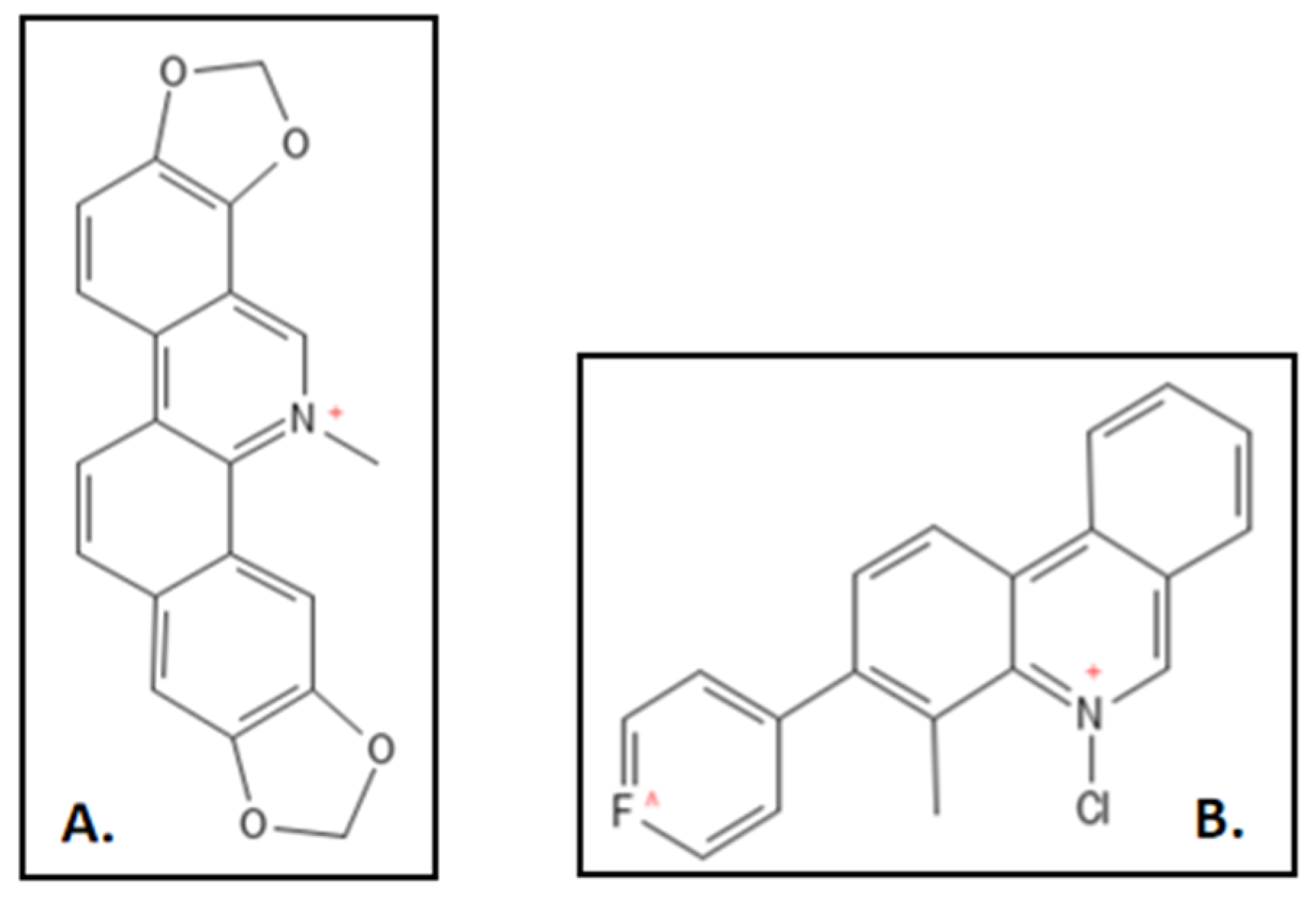
2. Sanguinarine
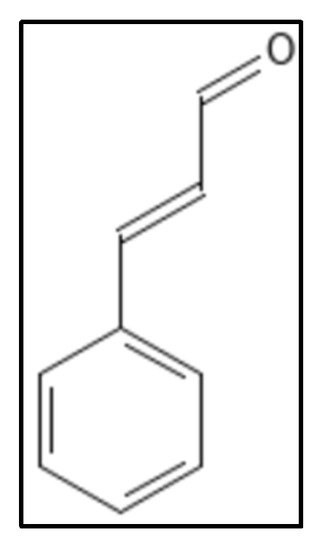
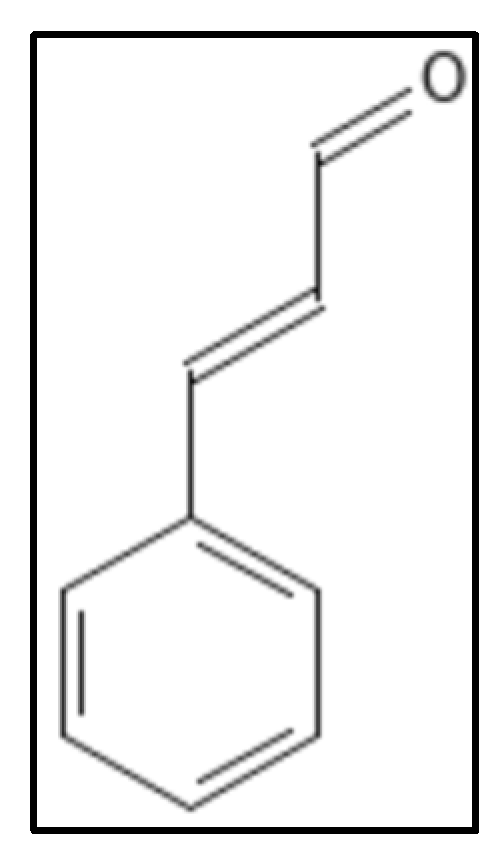
3. Cinnamaldehyde and Derivatives
4. Chrysophaentins
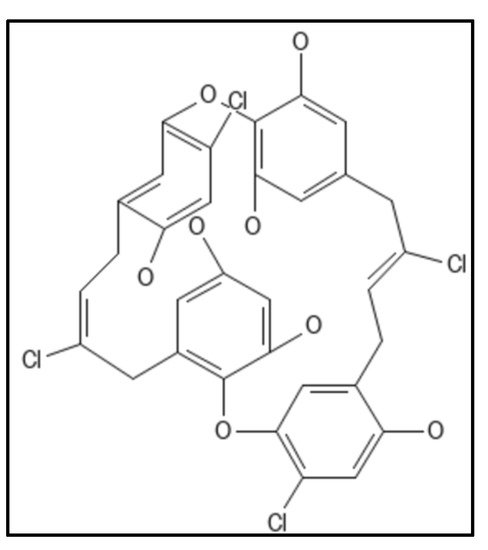
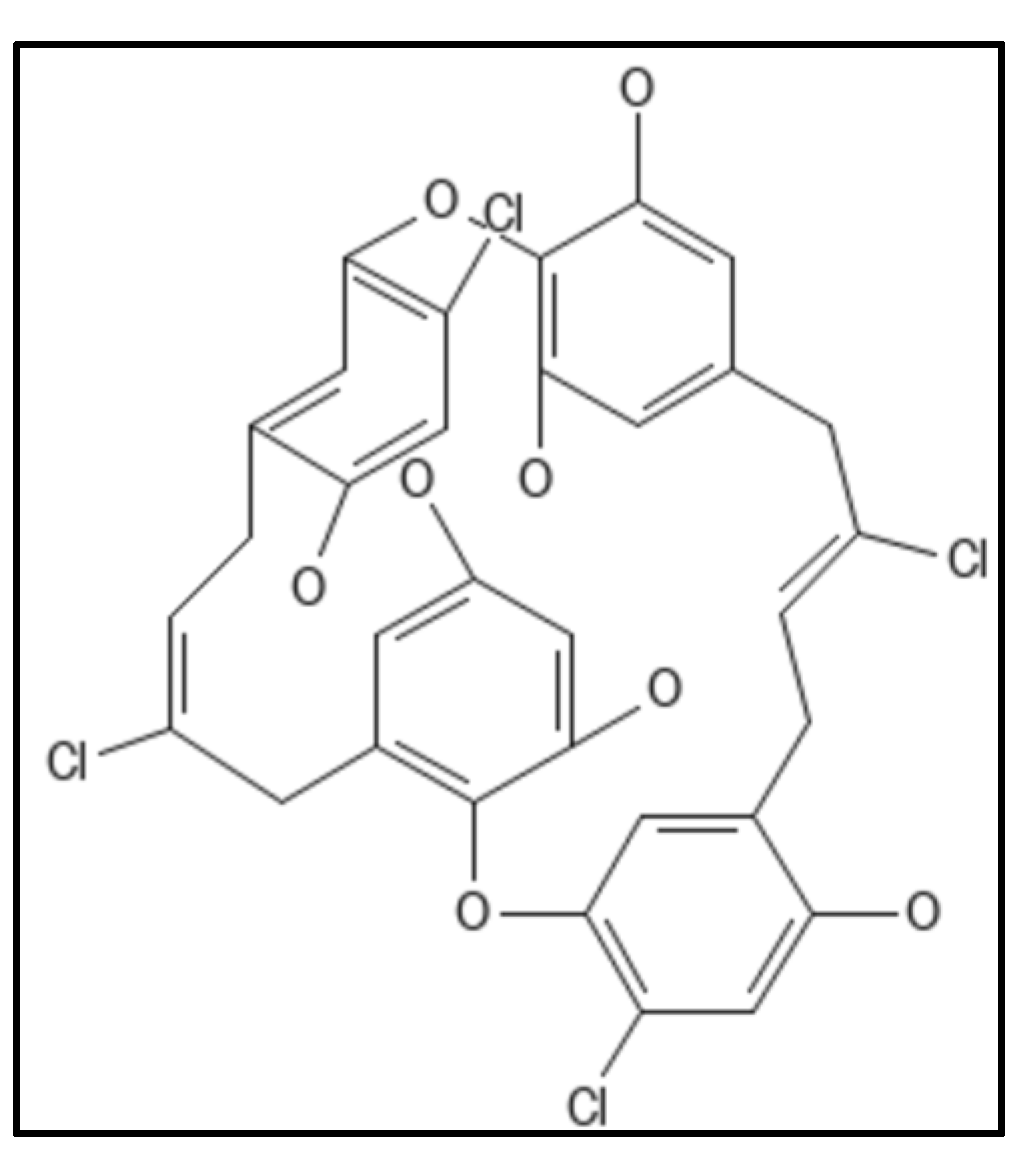
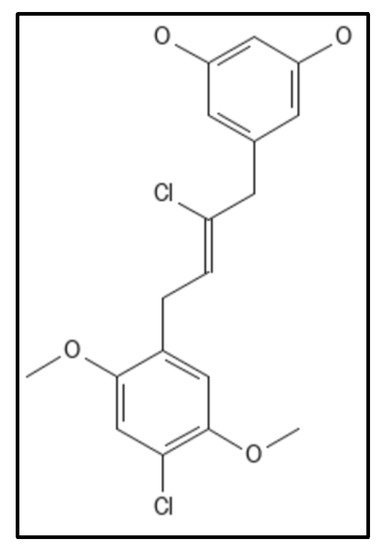
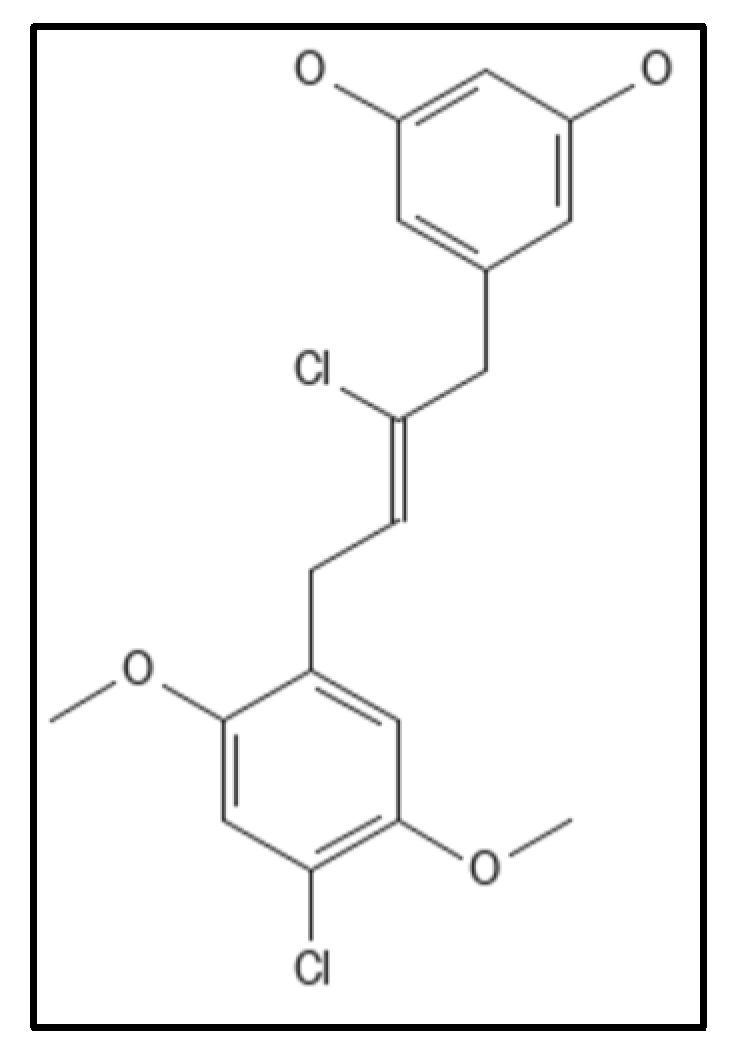
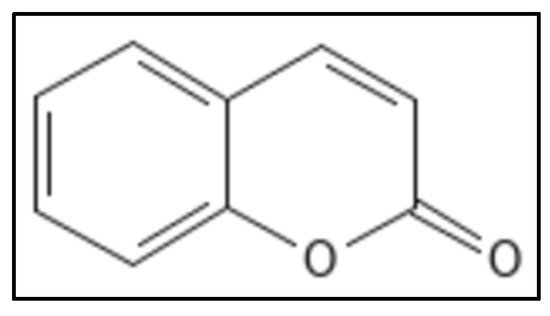
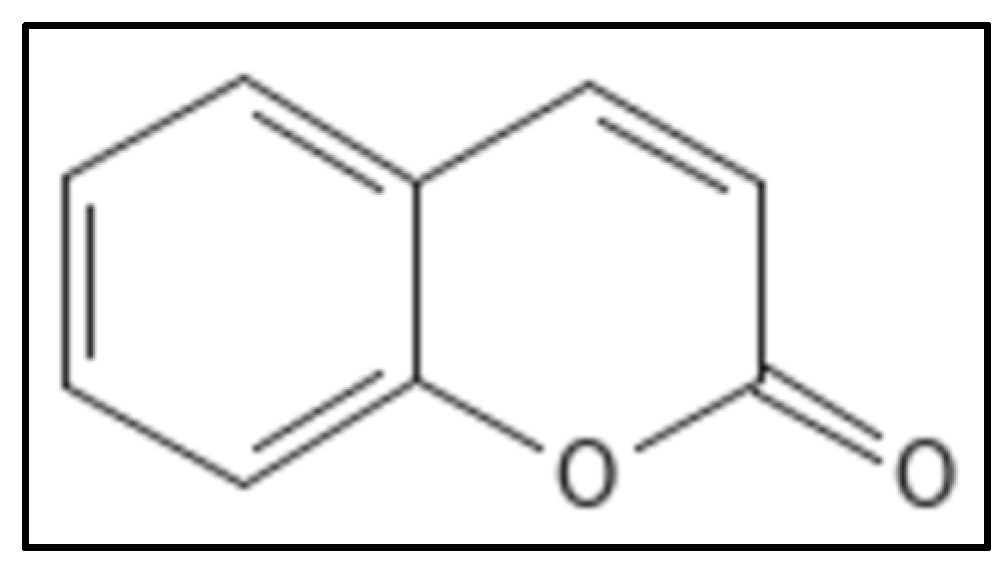
5. Coumarins
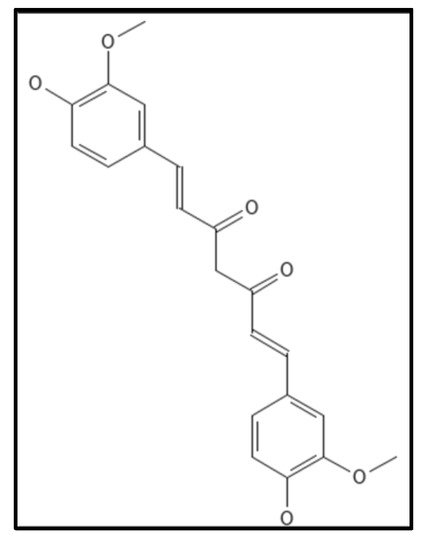
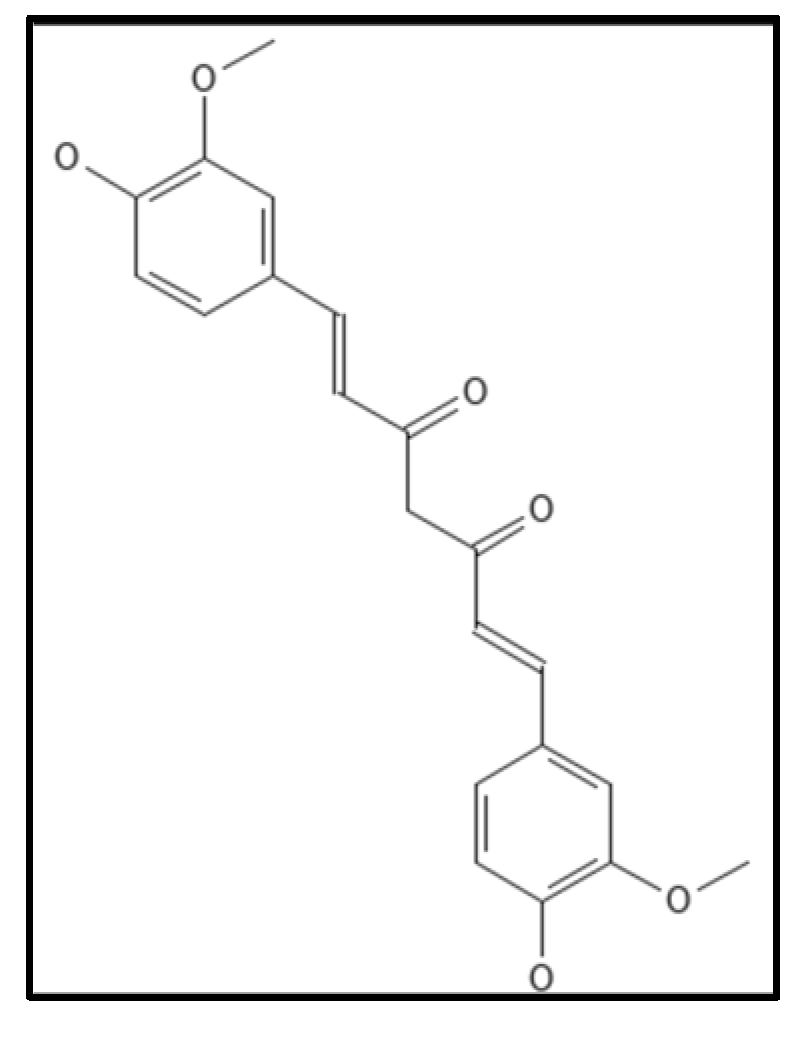
4.6. Curcumin
7. Dichamanetin and Derivatives
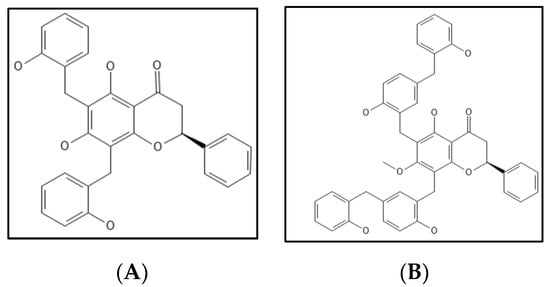
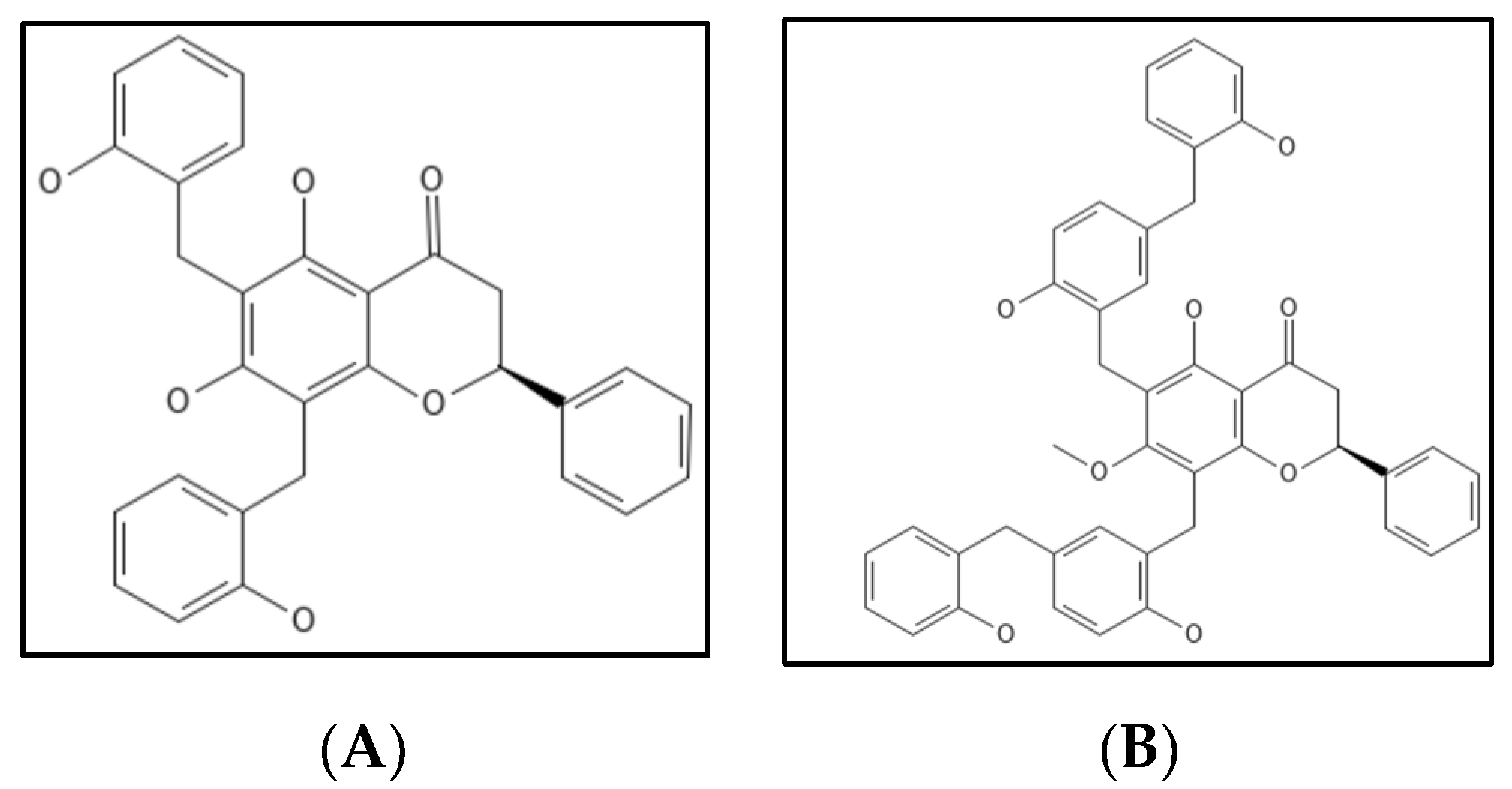
8. Doxorubicin
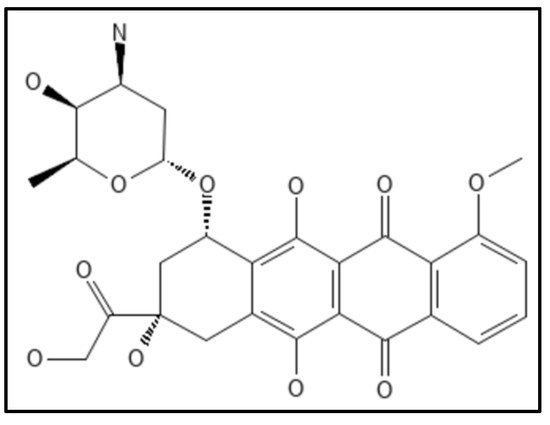
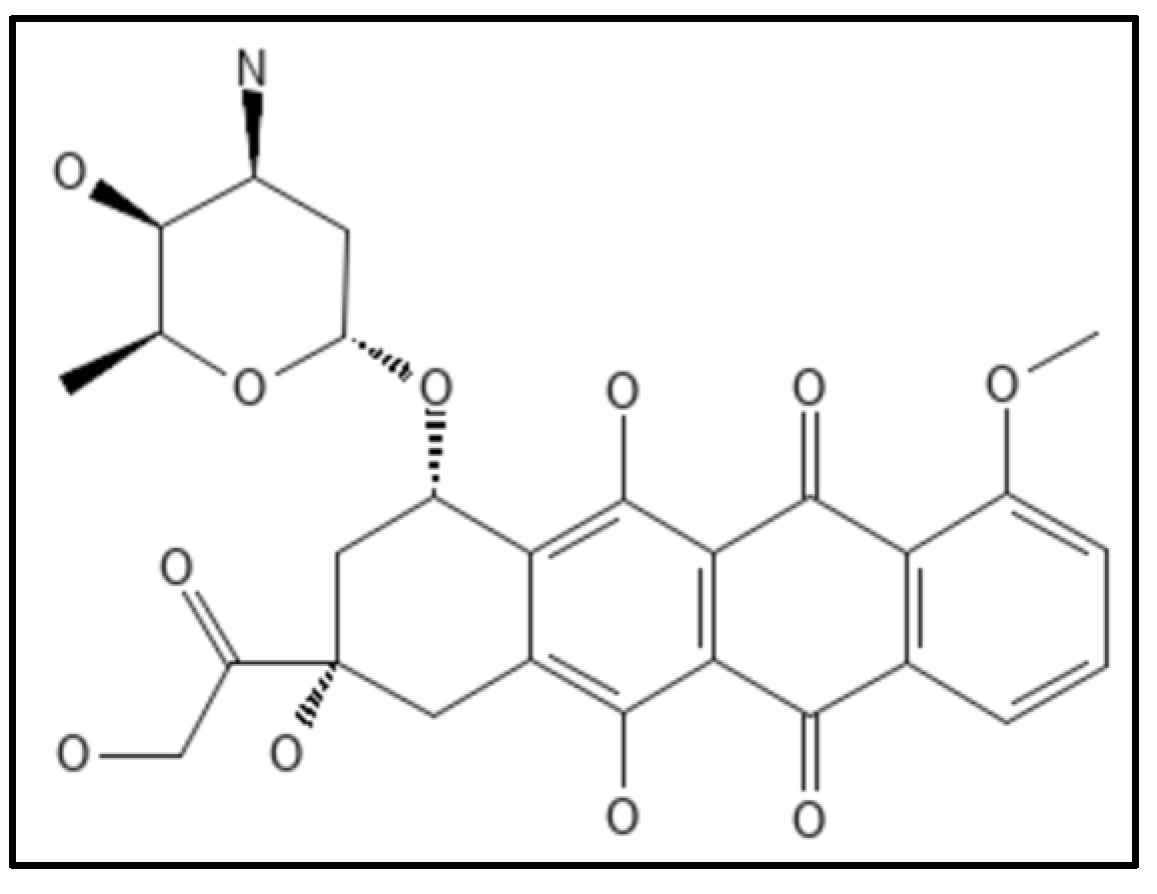
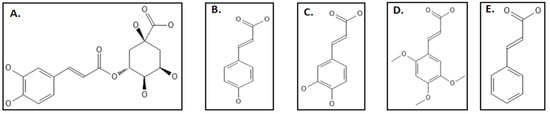
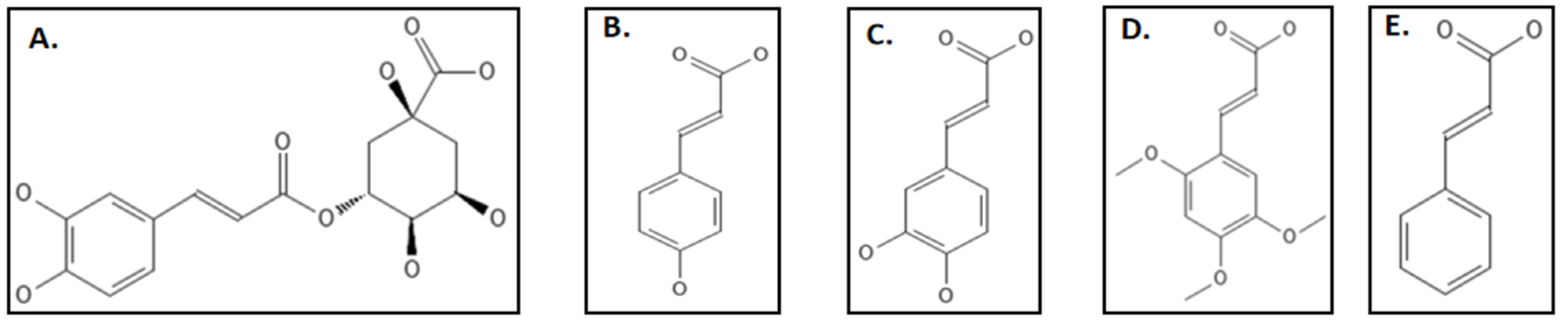
9. Phenylpropanoids
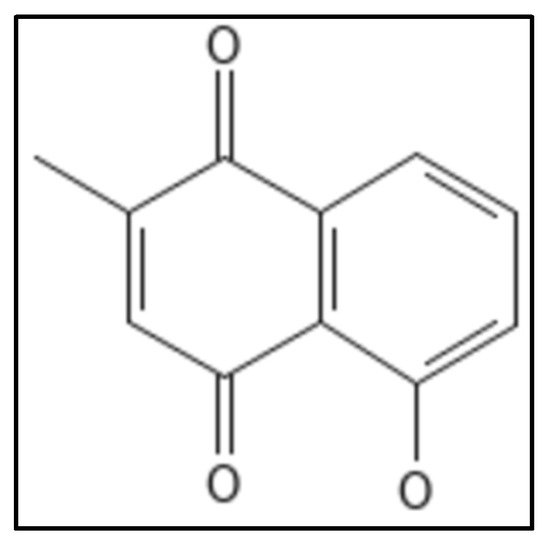
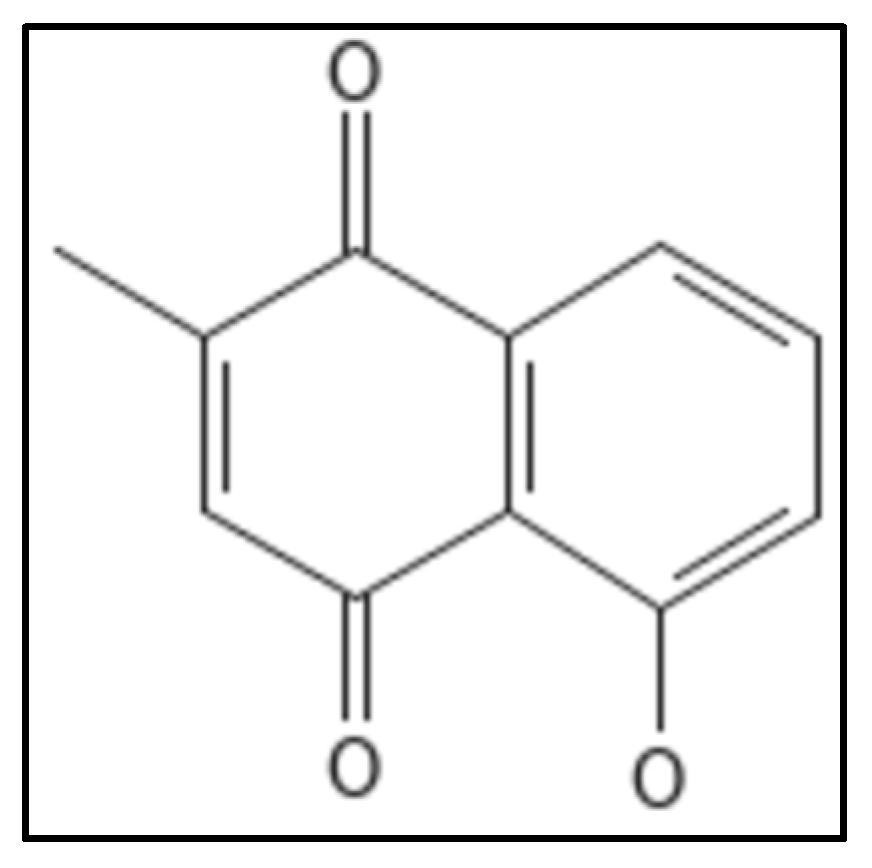
10. Plumbagin
11. Totarol
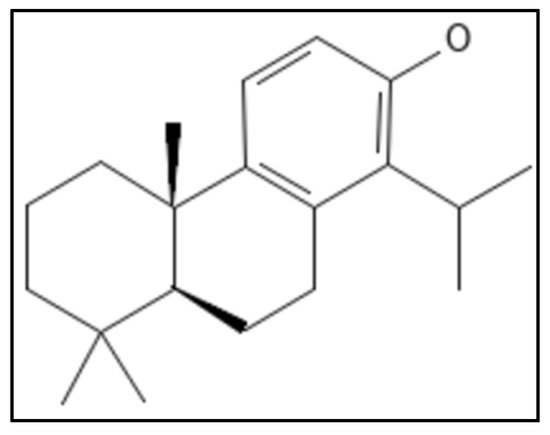
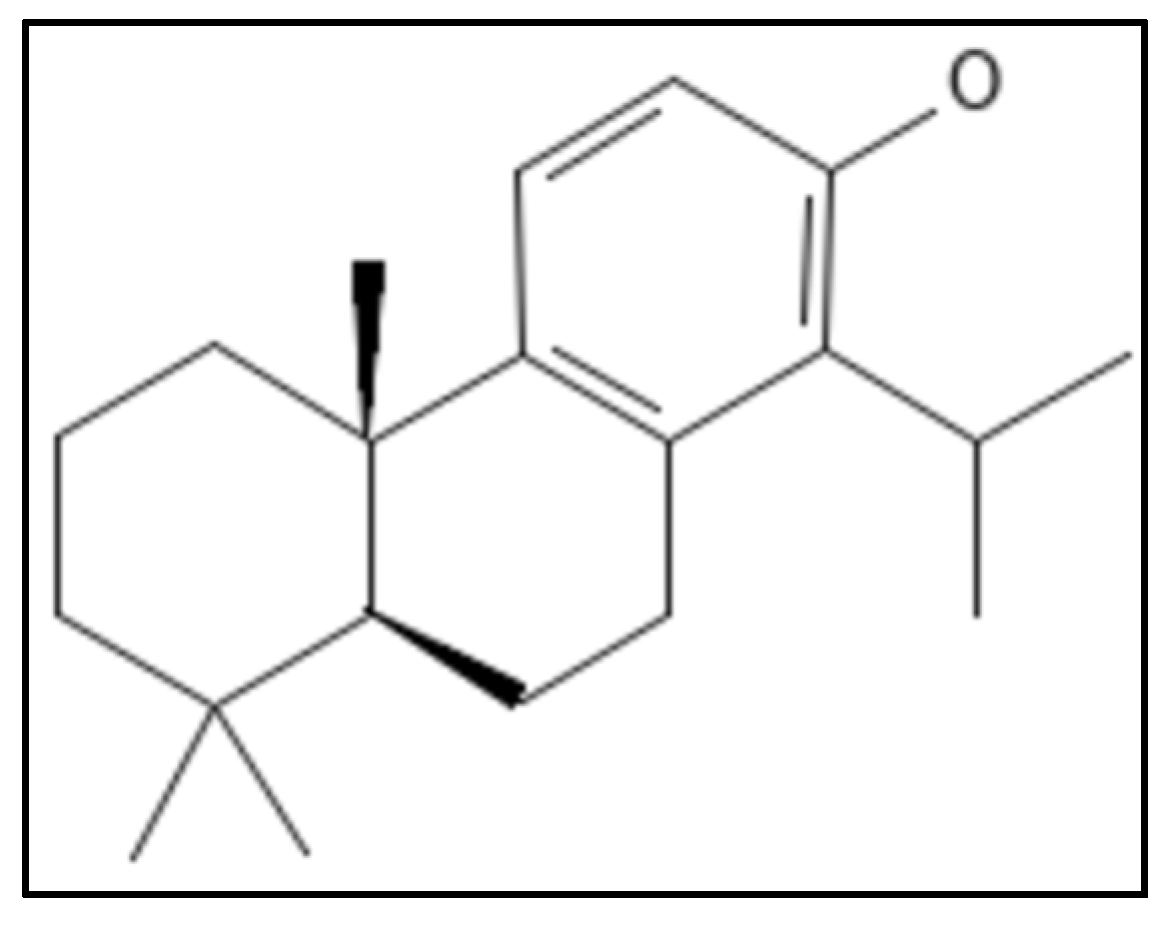
12. Viriditoxin
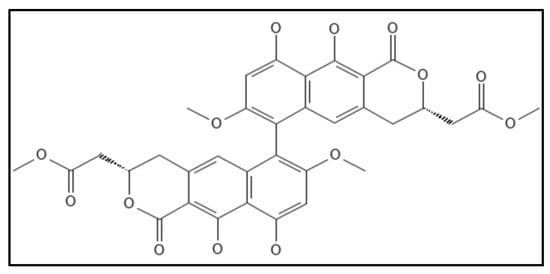
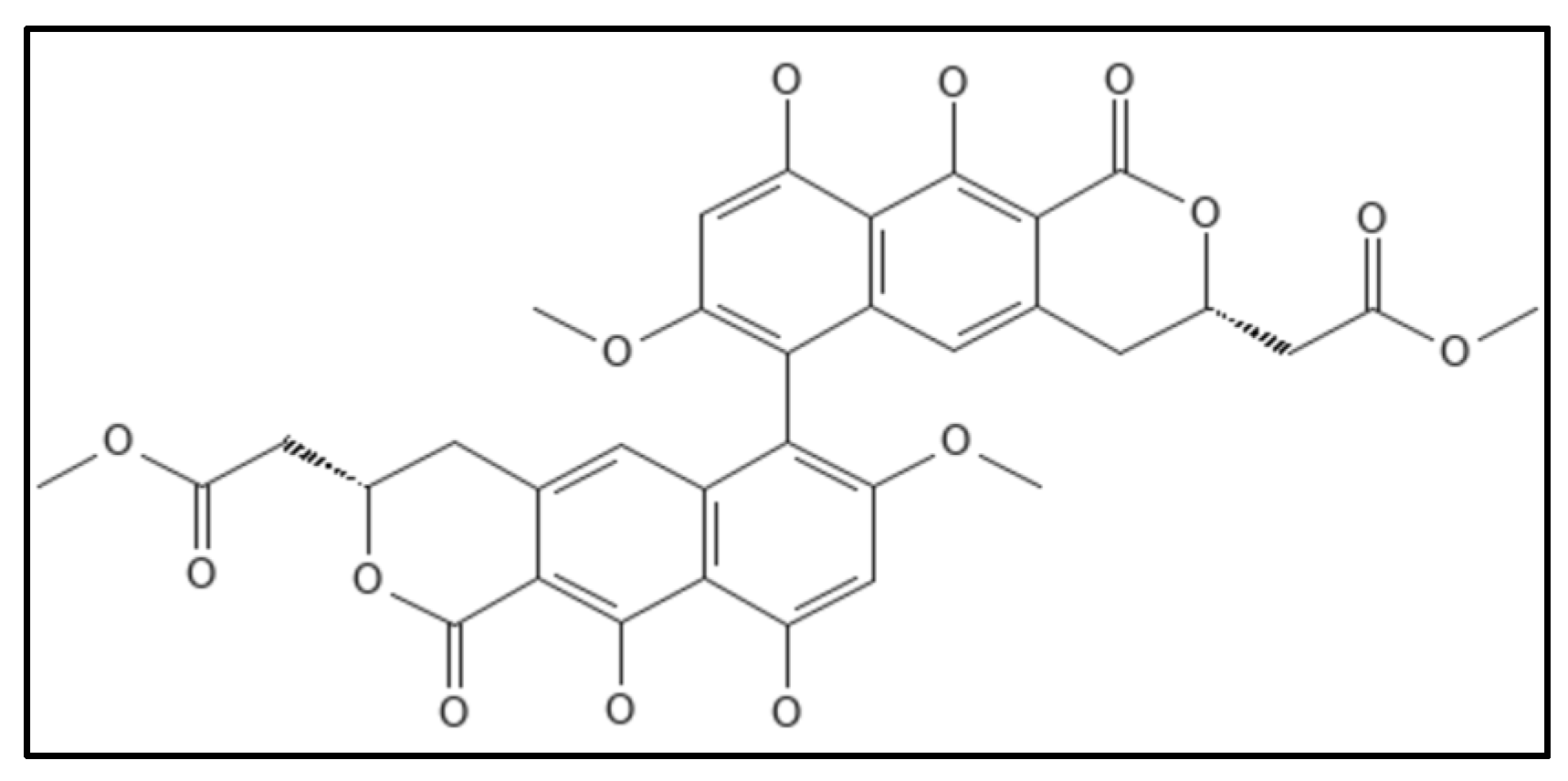
References
- Anca, I.-A.; Lumini, E.; Ghignone, S.; Salvioli, A.; Bianciotto, V.; Bonfante, P. The ftsZ Gene of the Endocellular Bacterium “Candidatus Glomeribacter gigasporarum” Is Preferentially Expressed during the Symbiotic Phases of Its Host Mycorrhizal Fungus. Mol. Plant Microbe Interact. MPMI 2009, 22, 302–310.
- Domadia, P.N.; Bhunia, A.; Sivaraman, J.; Swarup, S.; Dasgupta, D. Berberine targets assembly of Escherichia coli cell division protein FtsZ. Biochemistry 2008, 47, 3225–3234.
- Battaje, R.R.; Panda, D. Lessons from bacterial homolog of tubulin, FtsZ for microtubule dynamics. Endocr. Relat. Cancer 2017, 24, T1–T21.
- Sun, N.; Chan, F.Y.; Lu, Y.J.; Neves, M.A.C.; Lui, H.K.; Wang, Y.; Chow, K.Y.; Chan, K.F.; Yan, S.C.; Leung, Y.C.; et al. Rational design of berberine-based FtsZ inhibitors with broad-spectrum antibacterial activity. PLoS ONE 2014, 9, e97514.
- Cathie, I.A.B. Neonatal jaundice. Med. World 1947, 67, 104–106.
- Kumar, K.; Awasthi, D.; Berger, W.T.; Tonge, P.J.; Slayden, R.A.; Ojima, I. Discovery of anti-TB agents that target the cell-division protein FtsZ. Future Med. Chem. 2010, 2, 1305–1323.
- Beuria, T.K.; Santra, M.K.; Panda, D. Sanguinarine blocks cytokinesis in bacteria by inhibiting FtsZ assembly a bundling. Biochemistry 2005, 44, 16584–16593.
- Shreaz, S.; Wani, W.A.; Behbehani, J.M.; Raja, V.; Irshad, M.; Karched, M.; Ali, I.; Siddiqi, W.A.; Hun, L.T. Cinnamaldehyde and its derivatives, a novel class of antifungal agents. Fitoterapia 2016, 112, 116–131.
- Haranahalli, K.; Tong, S.; Ojima, I. Recent advances in the discovery and development of antibacterial agents targeting the cell-division protein FtsZ. Bioorg. Med. Chem. 2016, 24, 6354–6369.
- Domadia, P.; Swarup, S.; Bhunia, A.; Sivaraman, J.; Dasgupta, D. Inhibition of bacterial cell division protein FtsZ by cinnamaldehyde. Biochem. Pharmacol. 2007, 74, 831–840.
- Li, X.; Sheng, J.; Huang, G.; Ma, R.; Yin, F.; Song, D.; Zhao, C.; Ma, S. Design, synthesis and antibacterial activity of cinnamaldehyde derivatives as inhibitors of the bacterial cell division protein FtsZ. Eur. J. Med. Chem. 2015, 97, 32–41.
- Maier, S.K.; Scherer, S.; Loessner, M.J. Long-chain polyphosphate causes cell lysis and inhibits Bacillus cereus septum formation, which is dependent on divalent cations. Appl. Environ. Microbiol. 1999, 65, 3942–3949.
- Keffer, J.L.; Bifulco, G.; Lloyd, J.R.; Bewley, C.A.; Huecas, S.; Hammill, J.T.; Wipf, P.; Andreu, J.M.; Bewley, C.A. Chrysophaentins are competitive inhibitors of FtsZ and inhibit Z-ring formation in live bacteria. J. Am. Chem. Soc. 2013, 132, 5673–5678.
- Margolin, W.; Krupka, M. Unite to divide: Oligomerization of tubulin and actin homologs regulates initiation of bacterial cell division. F1000Research 2018, 7, 235.
- Liao, Y.; Ithurbide, S.; Löwe, J.; Duggin, I.G. Two FtsZ proteins orchestrate archaeal cell division through distinct functions in ring assembly and constriction. bioRxiv 2020.
- Pilch, D.S.; Lavoie, E.J.; Kaul, M.; Parhi, A. Benzo Phenanthridines as Antimicrobial Agents. U.S. Patent 8,741,917, 2010.
- Musa, M.A.; Badisa, V.L.D.; Latinwo, L.M.; Cooperwood, J.; Sinclair, A.; Abdullah, A. Cytotoxic activity of new acetoxycoumarin derivatives in cancer cell lines. Anticancer Res. 2011, 31, 2017–2022.
- Matos, M.J.; Vazquez-Rodriguez, S.; Santana, L.; Uriarte, E.; Fuentes-Edfuf, C.; Santos, Y.; Muñoz-Crego, A. Synthesis and Structure-Activity Relationships of Novel Amino/Nitro Substituted 3-Arylcoumarins as Antibacterial Agents. Molecules 2013, 18, 1394.
- Liu, J.; Ma, R.; Bi, F.; Zhang, F.; Hu, C.; Venter, H.; Semple, S.J.; Ma, S. Novel 5-methyl-2-phenylphenanthridium derivatives as FtsZ-targeting antibacterial agents from structural simplification of natural product sanguinarine. Bioorg. Med. Chem. Lett. 2018, 28, 1825–1831.
- Kumar, N.; Ashaq, M. Safety and Toxicity of Botanical Medicines: A critical Appraisal. Int. J. All Res. Educ. Sci. Methods 2021, 9, 2455–6211.
- Mitra, K.; Chadha, A.; Doble, M. Pharmacophore based approach to screen and evaluate novel Mycobacterium cell division inhibitors targeting FtsZ-A modelling and experimental study. Eur. J. Pharm. Sci. 2019, 135, 103–112.
- Riveiro, M.; De Kimpe, N.; Moglioni, A.; Vazquez, R.; Monczor, F.; Shayo, C.; Davio, C. Coumarins: Old compounds with novel promising therapeutic perspectives. Curr. Med. Chem. 2010, 17, 1325–1338.
- Sridevi, D.; Sudhakar, K.U.; Ananthathatmula, R.; Nankar, R.P.; Doble, M. Mutation at G103 of MtbFtsZ altered their sensitivity to coumarins. Front. Microbiol. 2017, 8, 578.
- Duggirala, S.; Nankar, R.; Rajendran, S. Phytochemicals as Inhibitors of Bacterial Cell Division Protein FtsZ: Coumarins Are Promising Candidates. Appl. Biochem. Biotechnol. 2014, 174, 283–296.
- Chattopadhyay, I.; Biswas, K.; Bandyopadhyay, U.; Banerjee, R.K. Turmeric and curcumin: Biological actions and medicinal applications. Curr. Sci. 2004, 87, 44–53.
- Alnami, A.; Norton, R.S.; Pena, H.P.; Haider, S.; Kozielski, F. Conformational Flexibility of A Highly Conserved Helix Controls Cryptic Pocket Formation in FtsZ. J. Mol. Biol. 2021, 433, 167061.
- Kaur, S.; Modi, N.H.; Panda, D.; Roy, N. Probing the binding site of curcumin in Escherichia coli and Bacillus subtilis FtsZ-A structural insight to unveil antibacterial activity of curcumin. Eur. J. Med. Chem. 2010, 45, 4209–4214.
- Sankhwar, R.; Yadav, S.; Kumar, A.; Gupta, R.K. Application of nano-curcumin as a natural antimicrobial agent against gram-positive pathogens. J. Appl. Nat. Sci. 2021, 13, 110–126.
- Urgaonkar, S.; La Pierre, H.S.; Meir, I.; Lund, H.; RayChaudhuri, D.; Shaw, J.T. Synthesis of antimicrobial natural products targeting FtsZ: (±)-dichamanetin and (±)-2‴-hydroxy-5″- benzylisouvarinol-B. Org. Lett. 2005, 7, 5609–5612.
- Panda, P.; Taviti, A.C.; Satpati, S.; Kar, M.M.; Dixit, A.; Beuria, T.K. Doxorubicin inhibits E. coli division by interacting at a novel site in FtsZ. Biochem. J. 2015, 471, 335–346.
- Hemaiswarya, S.; Doble, M. Synergistic interaction of phenylpropanoids with antibiotics against bacteria. J. Med. Microbiol. 2010, 59, 1469–1476.
- Li, X.; Sheng, J.; Song, D.; Guo, L.; Ma, S. Phenylacrylamides as Novel FtsZ-targeted Potential Antimicrobials. Lett. Drug Des. Discov. 2014, 12, 234–240.
- Sun, J.; Li, M.H.; Wang, X.Y.; Zhang, Y.; Yuan, R.J.; Liu, H.Y.; Zhu, H.L. Vanillin derivatives as the selective small molecule inhibitors of FtsZ. Med. Chem. Res. 2014, 23, 2985–2994.
- Sun, J.; Yin, Y.; Sheng, G.H.; Yang, Z.B.; Zhu, H.L. Synthesis, molecular modeling and structural characterization of vanillin derivatives as antimicrobial agents. J. Mol. Struct. 2013, 1039, 214–218.
- Mathew, R.; Kruthiventi, A.K.; Prasad, J.V.; Kumar, S.P.; Srinu, G.; Chatterji, D. Inhibition of mycobacterial growth by plumbagin derivatives. Chem. Biol. Drug Des. 2010, 76, 34–42.
- Ferreira, D.T.; Andrei, C.C.; Saridakis, H.O.; De Jesus Faria, T.; Vinhato, E.; Carvalho, K.E.; Daniel, J.F.S.; Machado, S.L.; Saridakis, D.P.; Braz-Filho, R. Antimicrobial activity and chemical investigation of Brazilian Drosera. Mem. Inst. Oswaldo Cruz 2004, 99, 753–755.
- Bhattacharya, A.; Jindal, B.; Singh, P.; Datta, A.; Panda, D. Plumbagin inhibits cytokinesis in Bacillus subtilis by inhibiting FtsZ assembly—A mechanistic study of its antibacterial activity. FEBS J. 2013, 280, 4585–4599.
- De Paiva, S.R.; Figueiredo, M.R.; Aragão, T.V.; Coelho Kaplan, M.A. Antimicrobial Activity in Vitro of Plumbagin Isolated from Plumbago Species. Mem. Inst. Oswaldo Cruz 2003, 98, 959–961.
- Acharya, B.R.; Bhattacharyya, B.; Chakrabarti, G. The natural naphthoquinone plumbagin exhibits antiproliferative activity and disrupts the microtubule network through tubulin binding. Biochemistry 2008, 47, 7838–7845.
- Jaiswal, R.; Beuria, T.K.; Mohan, R.; Mahajan, S.K.; Panda, D. Totarol inhibits bacterial cytokinesis by perturbing the assembly dynamics of FtsZ. Biochemistry 2007, 46, 4211–4220.
- Kim, M.B.; Shaw, J.T. Synthesis of antimicrobial natural products targeting FtsZ: (+)-totarol and related totarane diterpenes. Org. Lett. 2010, 12, 3324–3327.
- Su, M.; Zhao, C.; Li, D.; Cao, J.; Ju, Z.; Kim, E.L.; Jung, Y.S.; Jung, J.H. Viriditoxin stabilizes microtubule polymers in SK-OV-3 cells and exhibits antimitotic and antimetastatic potential. Mar. Drugs 2020, 18, 445.
- Wang, J.; Galgoci, A.; Kodali, S.; Herath, K.B.; Jayasuriya, H.; Dorso, K.; Vicente, F.; González, A.; Cully, D.; Bramhill, D.; et al. Discovery of a Small Molecule that Inhibits Cell Division by Blocking FtsZ, a Novel Therapeutic Target of Antibiotics. J. Biol. Chem. 2003, 278, 44424–44428.