Pyroptosis is a gasdermin-mediated, membrane pore forming, proinflammatory type of cell death. This necrotic form of cell death causes cell swelling and lysis and was initially found to be an infection triggered event occurring mostly in myeloid cells which leads to ion fluxes and release of proteins of the interleukin (IL) family. It was first described in 1992 by a group that saw a Caspase 1-dependent form of cell death in macrophages that were infected with Shigella flexneri and initially thought it was a form of apoptosis because of its caspase dependency. Pyroptosis is an important event in non-infectious diseases as well and therapeutic approaches have been developed in the hope of influencing several of these disease outcomes. The role of pyroptosis is described in sterile inflammation diseases, neuronal disease, cancer, atherosclerosis, autoimmune disease, acute injury, and adverse pregnancy events.
- pyroptosis
- gasdermins
- NLRP3
- SARS-CoV-2
- interleukin-1β
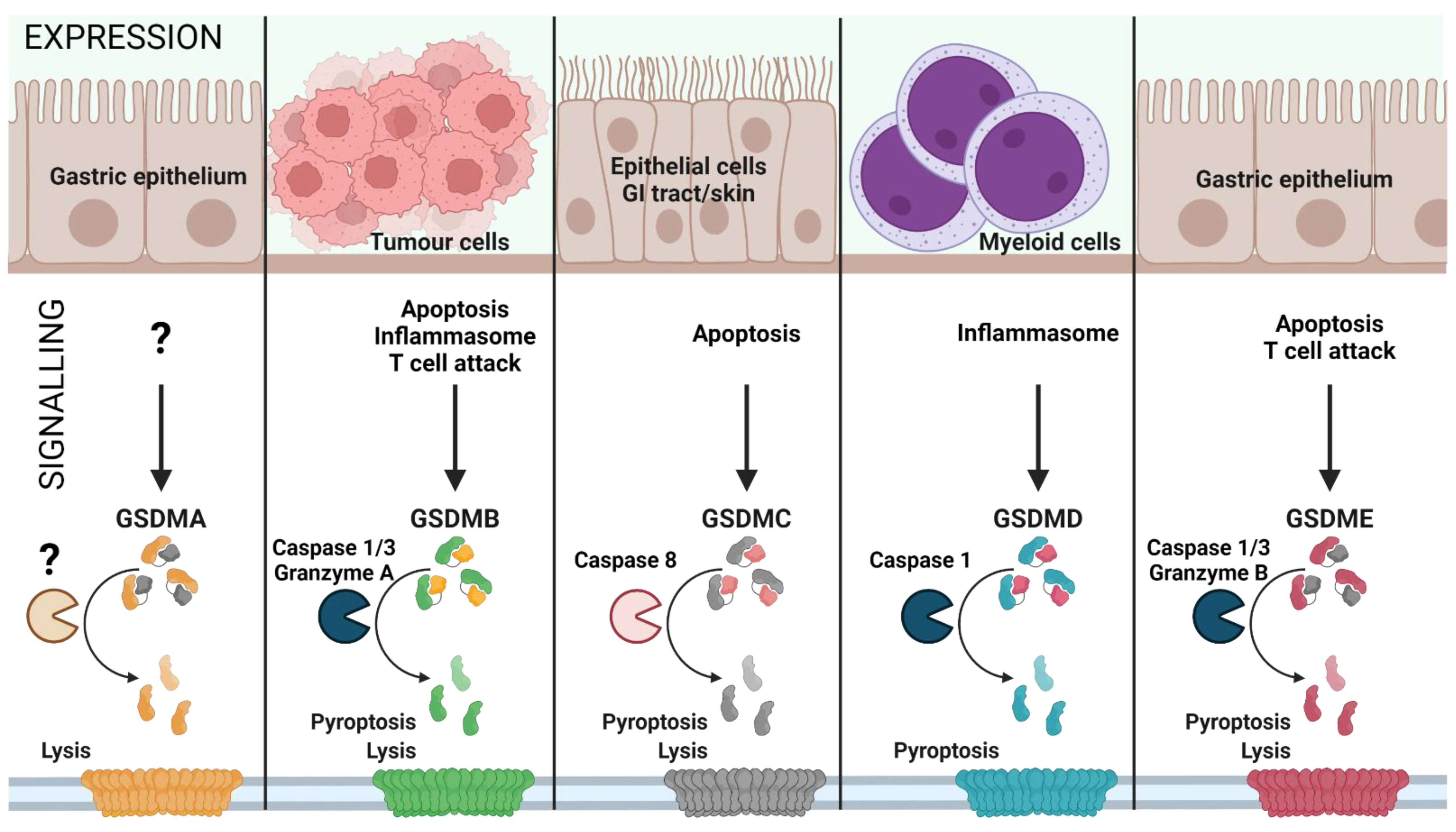
1. GSDMs as Therapeutic Targets
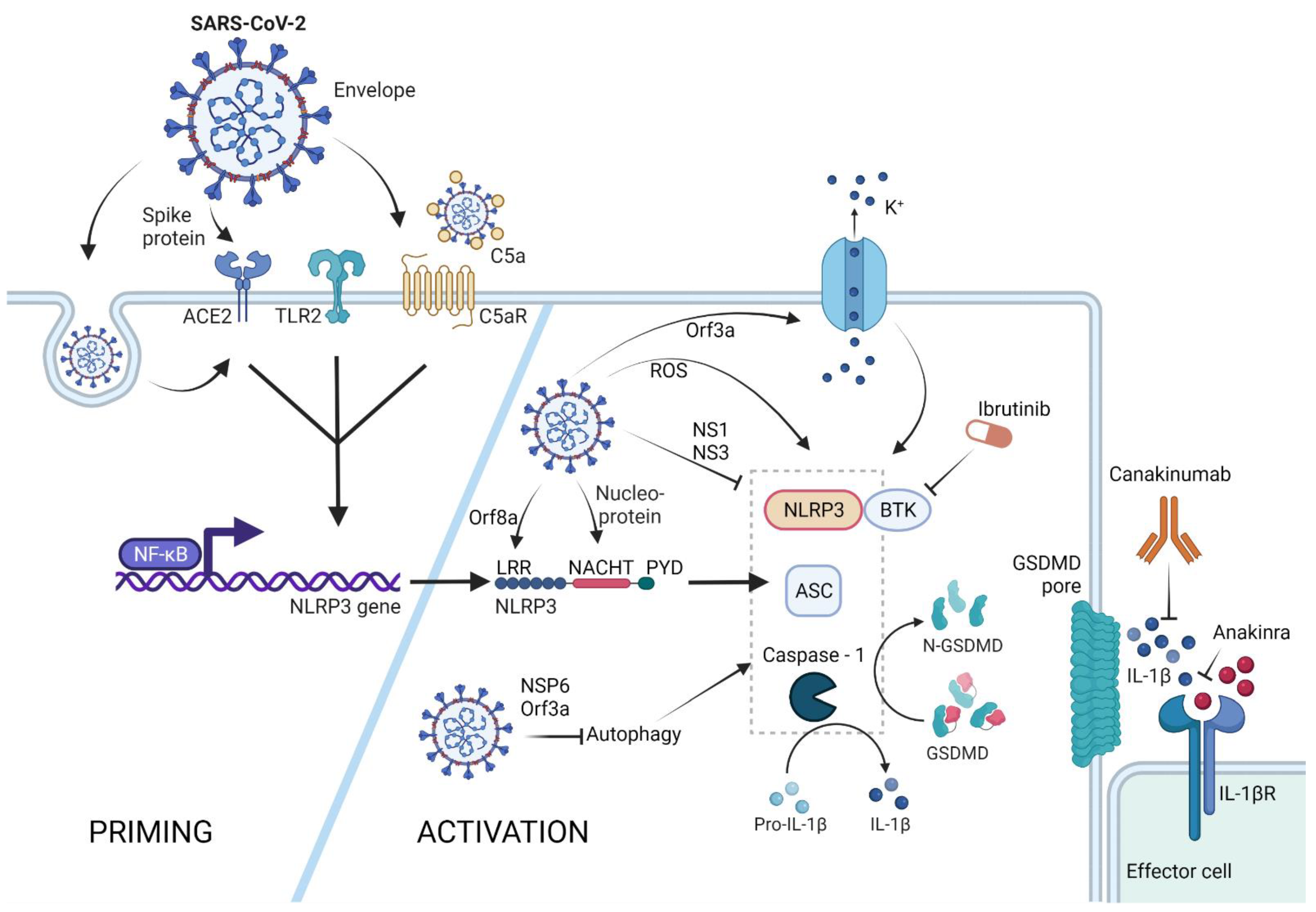
2. SARS-CoV-2 Triggered NLRP3-Mediated Pyroptosis
References
- Rathkey, J.K.; Zhao, J.; Liu, Z.; Chen, Y.; Yang, J.; Kondolf, H.C.; Benson, B.L.; Chirieleison, S.M.; Huang, A.Y.; Dubyak, G.R.; et al. Chemical disruption of the pyroptotic pore-forming protein gasdermin D inhibits inflammatory cell death and sepsis. Sci. Immunol. 2018, 3, eaat2738.
- Hu, J.J.; Liu, X.; Xia, S.; Zhang, Z.; Zhang, Y.; Zhao, J.; Ruan, J.; Luo, X.; Lou, X.; Bai, Y.; et al. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat. Immunol. 2020, 21, 736–745.
- Martín-Sánchez, F.; Diamond, C.; Zeitler, M.; Gomez, A.I.; Baroja-Mazo, A.; Bagnall, J.; Spiller, D.; White, M.; Daniels, M.; Mortellaro, A.; et al. Inflammasome-dependent IL-1β release depends upon membrane permeabilisation. Cell Death Differ. 2016, 23, 1219–1231.
- Juliana, C.; Fernandes-Alnemri, T.; Wu, J.; Datta, P.; Solorzano, L.; Yu, J.-W.; Meng, R.; Quong, A.A.; Latz, E.; Scott, C.P.; et al. Anti-inflammatory Compounds Parthenolide and Bay 11-7082 Are Direct Inhibitors of the Inflammasome. J. Biol. Chem. 2010, 285, 9792–9802.
- Mitra, S.; Exline, M.; Habyarimana, F.; Gavrilin, M.; Baker, P.; Masters, S.L.; Wewers, M.D.; Sarkar, A. Microparticulate Caspase 1 Regulates Gasdermin D and Pulmonary Vascular Endothelial Cell Injury. Am. J. Respir. Cell Mol. Biol. 2018, 59, 56–64.
- Humphries, F.; Shmuel-Galia, L.; Ketelut-Carneiro, N.; Li, S.; Wang, B.; Nemmara, V.V.; Wilson, R.; Jiang, Z.; Khalighinejad, F.; Muneeruddin, K.; et al. Succination inactivates gasdermin D and blocks pyroptosis. Science 2020, 369, 1633–1637.
- Diaz, G.M.; Hupperts, R.; Fraussen, J.; Somers, V. Dimethyl fumarate treatment in multiple sclerosis: Recent advances in clinical and immunological studies. Autoimmun. Rev. 2018, 17, 1240–1250.
- Hu, L.; Chen, M.; Chen, X.; Zhao, C.; Fang, Z.; Wang, H.; Dai, H. Chemotherapy-induced pyroptosis is mediated by BAK/BAX-caspase-3-GSDME pathway and inhibited by 2-bromopalmitate. Cell Death Dis. 2020, 11, 281.
- Hergueta-Redondo, M.; Sarrio, D.; Molina-Crespo, A.; Vicario, R.; Morales, C.B.; Martínez, L.; Rojo-Sebastián, A.; Serra-Musach, J.; Mota, A.; Martínez-Ramírez, A.; et al. Gasdermin B expression predicts poor clinical outcome in HER2-positive breast cancer. Oncotarget 2016, 7, 56295–56308.
- Molina-Crespo, A.; Cadete, A.; Sarrio, D.; Gamez-Chiachio, M.; Martinez, L.; Chao, K.; Olivera, A.; Gonella, A.; Diaz, E.; Palacio, H.; et al. Intracellular delivery of an antibody targeting gasdermin-b reduces her2 breast cancer aggressiveness. Clin. Cancer Res. 2019, 25, 4846–4858.
- Chan, J.F.-W.; Kok, K.-H.; Zhu, Z.; Chu, H.; To, K.K.-W.; Yuan, S.; Yuen, K.-Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020, 9, 221–236.
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034.
- Toldo, S.; Bussani, R.; Nuzzi, V.; Bonaventura, A.; Mauro, A.G.; Cannatà, A.; Pillappa, R.; Sinagra, G.; Nana-Sinkam, P.; Sime, P.; et al. Inflammasome formation in the lungs of patients with fatal COVID-19. Agents Actions 2021, 70, 7–10.
- Rodrigues, T.S.; de Sa, K.S.G.; Ishimoto, A.Y.; Becerra, A.; Oliveira, S.; Almeida, L.; Goncalves, A.V.; Perucello, D.B.; Andrade, W.A.; Castro, R.; et al. Inflammasomes are activated in response to SARS-cov-2 infection and are associated with COVID-19 severity in patients. J. Exp. Med. 2020, 218, e20201707.
- Zeng, J.; Xie, X.; Feng, X.-L.; Xu, L.; Han, J.-B.; Yu, D.; Zou, Q.-C.; Liu, Q.; Li, X.; Ma, G.; et al. Specific inhibition of the NLRP3 inflammasome suppresses immune overactivation and alleviates COVID-19 like pathology in mice. eBioMedicine 2021, 75, 103803.
- Coll, R.C.; Hill, J.R.; Day, C.J.; Zamoshnikova, A.; Boucher, D.; Massey, N.L.; Chitty, J.; Fraser, J.A.; Jennings, M.P.; Robertson, A.A.B.; et al. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat. Chem. Biol. 2019, 15, 556–559.
- Ferreira, A.C.; Soares, V.C.; de Azevedo-Quintanilha, I.G.; Dias, S.d.S.G.; Fintelman-Rodrigues, N.; Sacramento, C.Q.; Mattos, M.; de Freitas, C.S.; Temerozo, J.R.; Teixeira, L.; et al. Correction: SARS-CoV-2 engages inflammasome and pyroptosis in human primary monocytes. Cell Death Discov. 2021, 7, 43.
- Landi, L.; Ravaglia, C.; Russo, E.; Cataleta, P.; Fusari, M.; Boschi, A.; Giannarelli, D.; Facondini, F.; Valentini, I.; Panzini, I.; et al. Blockage of interleukin-1β with canakinumab in patients with Covid-19. Sci. Rep. 2020, 10, 21775.
- Grein, J.; Ohmagari, N.; Shin, D.; Diaz, G.; Asperges, E.; Castagna, A.; Feldt, T.; Green, G.; Green, M.L.; Lescure, F.X.; et al. Compassionate Use of Remdesivir for Patients with Severe Covid-19. N. Engl. J. Med. 2020, 382, 2327–2336.
- Cavalli, G.; de Luca, G.; Campochiaro, C.; Della-Torre, E.; Ripa, M.; Canetti, D.; Oltolini, C.; Castiglioni, B.; Din, C.T.; Boffini, N.; et al. Interleukin-1 blockade with high-dose anakinra in patients with COVID-19, acute respiratory distress syndrome, and hyperinflammation: A retrospective cohort study. Lancet Rheumatol. 2020, 2, e325–e331.
- Pontali, E.; Volpi, S.; Antonucci, G.; Castellaneta, M.; Buzzi, D.; Tricerri, F.; Angelelli, A.; Caorsi, R.; Feasi, M.; Calautti, F.; et al. Safety and efficacy of early high-dose IV anakinra in severe COVID-19 lung disease. J. Allergy Clin. Immunol. 2020, 146, 213–215.
- Pasin, L.; Cavalli, G.; Navalesi, P.; Sella, N.; Landoni, G.; Yavorovskiy, A.G.; Likhvantsev, V.V.; Zangrillo, A.; Dagna, L.; Monti, G. Anakinra for patients with COVID-19: A meta-analysis of non-randomized cohort studies. Eur. J. Intern. Med. 2021, 86, 34–40.
- Kyriazopoulou, E.; Poulakou, G.; Milionis, H.; Metallidis, S.; Adamis, G.; Tsiakos, K.; Fragkou, A.; Rapti, A.; Damoulari, C.; Fantoni, M.; et al. Early treatment of COVID-19 with anakinra guided by soluble urokinase plasminogen receptor plasma levels: A double-blind, randomized controlled phase 3 trial. Nat. Med. 2021, 27, 1752–1760.
- Zhang, J.; Wu, H.; Yao, X.; Zhang, D.; Zhou, Y.; Fu, B.; Wang, W.; Li, H.; Wang, Z.; Hu, Z.; et al. Pyroptotic macrophages stimulate the SARS-CoV-2-associated cytokine storm. Cell. Mol. Immunol. 2021, 18, 1305–1307.
- Siu, K.-L.; Yuen, K.-S.; Castano-Rodriguez, C.; Ye, Z.-W.; Yeung, M.-L.; Fung, S.-Y.; Yuan, S.; Chan, C.-P.; Yuen, K.-Y.; Enjuanes, L.; et al. Severe acute respiratory syndrome Coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC. FASEB J. 2019, 33, 8865–8877.
- Chen, I.-Y.; Moriyama, M.; Chang, M.-F.; Ichinohe, T. Severe Acute Respiratory Syndrome Coronavirus Viroporin 3a Activates the NLRP3 Inflammasome. Front. Microbiol. 2019, 10, 50.
- Xu, H.; Chitre, S.A.; Akinyemi, I.A.; Loeb, J.C.; Lednicky, J.A.; McIntosh, M.T.; Bhaduri-McIntosh, S. SARS-CoV-2 viroporin triggers the NLRP3 inflammatory pathway. bioRxiv 2020, in press.
- Shi, C.-S.; Nabar, N.R.; Huang, N.-N.; Kehrl, J.H. SARS-Coronavirus Open Reading Frame-8b triggers intracellular stress pathways and activates NLRP3 inflammasomes. Cell Death Discov. 2019, 5, 101.
- Pan, P.; Shen, M.; Yu, Z.; Ge, W.; Chen, K.; Tian, M.; Xiao, F.; Wang, Z.; Wang, J.; Jia, Y.; et al. SARS-CoV-2 N protein promotes NLRP3 inflammasome activation to induce hyperinflammation. Nat. Commun. 2021, 12, 4664.
- Sun, X.; Liu, Y.; Huang, Z.; Xu, W.; Hu, W.; Yi, L.; Liu, Z.; Chan, H.; Zeng, J.; Liu, X.; et al. SARS-CoV-2 non-structural protein 6 triggers NLRP3-dependent pyroptosis by targeting ATP6AP1. Cell Death Differ. 2022, in press.
- Zheng, M.; Karki, R.; Williams, E.P.; Yang, D.; Fitzpatrick, E.; Vogel, P.; Jonsson, C.B.; Kanneganti, T.-D. TLR2 senses the SARS-CoV-2 envelope protein to produce inflammatory cytokines. Nat. Immunol. 2021, 22, 829–838.
- Cohen, M.E.; Eichel, R.; Steiner-Birmanns, B.; Janah, A.; Ioshpa, M.; Bar-Shalom, R.; Paul, J.J.; Gaber, H.; Skrahina, V.; Bornstein, N.M.; et al. A case of probable Parkinson’s disease after SARS-CoV-2 infection. Lancet Neurol. 2020, 19, 804–805.
- Albornoz, E.; Amarilla, A.A.; Modhiran, N.; Parker, S.; Li, X.X.; Wijesundara, D.K.; Zamora, A.P.; McMillan, C.L.D.; Liang, B.; Peng, N.Y.G.; et al. SARS-CoV-2 drives NLRP3 inflammasome activation in human microglia through spike-ACE2 receptor interaction. bioRxiv 2022, in press.
- Theobald, S.J.; Simonis, A.; Georgomanolis, T.; Kreer, C.; Zehner, M.; Eisfeld, H.S.; Albert, M.-C.; Chhen, J.; Motameny, S.; Erger, F.; et al. Long-lived macrophage reprogramming drives spike protein-mediated inflammasome activation in COVID-19. EMBO Mol. Med. 2021, 13, e14150.
- Junqueira, C.; Crespo, A.; Ranjbar, S.; Ingber, J.; Parry, B.; Ravid, S.; de Lacerda, L.B.; Lewandrowski, M.; Clark, S.; Ho, F.; et al. SARS-CoV-2 infects blood monocytes to activate NLRP3 and AIM2 inflammasomes, pyroptosis and cytokine release. medRxiv 2021, in press.
- Yu, J.; Yuan, X.; Chen, H.; Chaturvedi, S.; Braunstein, E.M.; Brodsky, R.A. Direct activation of the alternative complement pathway by SARS-CoV-2 spike proteins is blocked by factor D Inhibition. Blood 2020, 136, 2080–2089.
- Bournazos, S.; Gupta, A.; Ravetch, J.V. The role of IgG Fc receptors in antibody-dependent enhancement. Nat. Rev. Immunol. 2020, 20, 633–643.
- Kim, N.E.; Kim, D.K.; Song, Y.J. Sars-cov-2 nonstructural proteins 1 and 13 suppress caspase-1 and the NLRP3 inflammasome activation. Microorganisms 2021, 9, 494.
- Maggiore, U. Colchicine Counteracting Inflammation in COVID-19 Pneumonia (ColCOVID-19). 2020. Available online: https://clinicaltrials.gov/show/NCT04322565 (accessed on 13 April 2022).
- Mehta, K.G.; Patel, T.; Chavda, P.D.; Patel, P. Efficacy and safety of colchicine in COVID-19: A meta-analysis of randomised controlled trials. RMD Open 2021, 7, e001746.
- Treon, S.P.; Castillo, J.J.; Skarbnik, A.P.; Soumerai, J.D.; Ghobrial, I.M.; Guerrera, M.L.; Meid, K.E.; Yang, G. The BTK inhibitor ibrutinib may protect against pulmonary injury in COVID-19–infected patients. Blood 2020, 135, 1912–1915.
- Roschewski, M.; Lionakis, M.S.; Sharman, J.P.; Roswarski, J.; Goy, A.; Monticelli, M.A.; Roshon, M.; Wrzesinski, S.H.; Desai, J.V.; Zarakas, M.A.; et al. Inhibition of Bruton tyrosine kinase in patients with severe COVID-19. Sci. Immunol. 2020, 5, eabd0110.
- Zheng, M.; Williams, E.P.; Malireddi, R.K.S.; Karki, R.; Banoth, B.; Burton, A.; Webby, R.; Channappanavar, R.; Jonsson, C.B.; Kanneganti, T.-D. Impaired NLRP3 inflammasome activation/pyroptosis leads to robust inflammatory cell death via caspase-8/RIPK3 during coronavirus infection. J. Biol. Chem. 2020, 295, 14040–14052.