1. GSDMs as Therapeutic Targets
With the elucidation of the effectors of the pyroptosis pathway and their role in disease, the search for inhibitors has become a highly investigated field. The majority of promising drugs such as disulfiram and necrosulfonamide (NSA) target reactive cysteine residues because of their critical role in cytosol gasdermin recruitment and pore formation
[1][70]. Hu et al. characterized disulfiram, an FDA-approved drug for treating alcohol addiction as a potent LPS-induced sepsis inhibitor in mice. At nanomolar concentrations, disulfiram covalently modifies human Cys191 in GSDMD to block pore formation while allowing IL-1β and GSDMD processing
[2][71]. Punicalagin, a polyphenol, probably prevents NT-GSDMD cytoplasmic membrane insertion through its antioxidative effect on reactive thiols
[3][72]. Upstream inhibition of GSDMD pore formation takes place through inhibitors such as Bay 11–7082, an identified inhibitor of NF-κB, or z-VAD-fmk, a Caspase 1 specific inhibitor
[4][5][73,74] Natural modified metabolites such as unsaturated dicarbonic acids dimethlylfumarate (DMF) inactivate GSDMD and GSDME by succination at Cys101 and Cys45. DMF use in the treatment of neurological disorders such as multiple sclerosis (MS) is currently being evaluated
[6][7][50,75]. 2-Bromopalmitate was found to inhibit the CT-GSDME palmitoylation and prevent chemotherapy-induced GSDME-mediated pyroptosis
[8][15].
Due to the existence of many reactive thiols in the inflammatory pathways, the unspecific nature of all these drugs could likely be an issue with regard to off-target effects. More studies have to be conducted to characterize the mode of action of these compounds in vivo.
It has been found that treatment failure of aggressive HER2 positive breast cancers is associated with the coamplification and coexpression of an
Erb2 neighbour gene, namely, the
GSDMB gene
[9][79]. An approach independent of small molecules to prevent pyroptosis was published by Molina-Crespo et al. where an anti-GSDMB antibody was intracellularly delivered into HER2 positive breast cancer cells, proving that protumour GSDMB functions such as migration, metastasis, and therapy resistance could be reduced
[10][76]. This promising approach should be further evaluated for translational potential. Although many attempts have been made to inhibit pyroptosis in order to treat inflammation, most approaches have not reached drug approval and the definition and inhibition of targets during pyroptosis needs further elucidation.
2. SARS-CoV-2 Triggered NLRP3-Mediated Pyroptosis
With the rapid worldwide spread of the novel coronavirus (SARS-CoV-2) in late 2019; the World Health Organization (WHO) declared a global emergency. Since then, the pandemic has had a significant toll on human health and the world economy. Globally, to date there have been 497,057,239 confirmed cases of COVID-19, including 6,179,104 deaths, reported to WHO [
https://covid19.who.int/ (accessed on 12 April 2022)].
SARS-CoV-2 is an enveloped RNA virus that is composed of several proteins: the nucleocapsid, the matrix, the envelope, and the spike
[11][80] . It is transmitted from person to person primarily through droplet and aerosol routes. COVID-19 manifests most commonly as a respiratory illness with hyperinflammation of the lung in patients with severe disease
[12][81] . Consequently, clinicians have started to investigate whether and how inflammasome activation and pyroptosis are linked to COVID-19 symptoms. Understanding the connection between SARS-CoV-2 infection and pyroptosis-mediated inflammation has been critical since the emergence of the pandemic, and even more so because an inflammasome and pyroptosis-mediated inflammatory signature could present an opportunity for therapeutic intervention in which pyroptosis-associated events are targeted. In fact, it has been shown that SARS-CoV-2 infection leads to NLRP3 inflammasome activation in vitro and in vivo
[13][14][82,83]. Inflammasome activation was also associated with COVID-19 severity
[13][14][82,83] and NLRP3 activation might even be a suitable predictor of COVID-19 disease severity and a potential therapeutic target. Indeed, inhibition of the NLRP3 inflammasome with the well-characterized NLRP3 inhibitory compound MCC950 reduced COVID-19 pathology in mice
[15][16][17][84,85,86].
Furthermore, experimental treatment of severe COVID-19 with canakinumab, an anti-IL-1β antibody, showed some beneficial effects early into the pandemic; similar to that of compassionate use of remdesivir, a viral polymerase inhibitor
[18][19][77,87] (see
Figure 12).
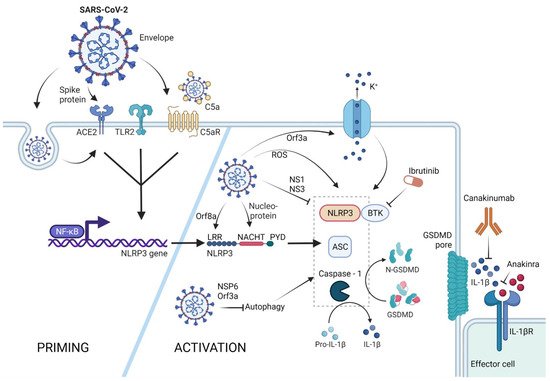
Figure 12. NLRP3 inflammasome activation by SARS-CoV-2. The NLRP3 inflammasome can be primed by SARS-CoV-2 proteins spike and envelope, and in its opsonized form by activating the ACE-2, TLR-2, and complement receptors, respectively. SARS-CoV-2-infected cells upregulate ACE-2 receptor expression, rendering these cells more sensitive to SARS-CoV-2 response. Activation of these receptors leads to NFκB-mediated Nlrp3 gene transcription and translation. The NLRP3 inflammasome can form in response to ROS generation in SARS-CoV-2-infected cells, via potassium efflux through SARS-CoV-2 Orf3a-generated pores, or via direct interaction with the SARS-CoV-2 proteins orf8a and nucleoprotein. SARS-CoV-2 is capable of NLRP3 inflammasome inhibition via NS1 and NS13 proteins, as well as through inhibition of autophagy, a general NLRP3 trigger. Therapeutic potential lies in the inhibition of the effector molecule IL-1β via canakinumab or anakinra, as well as inhibition of the NLRP3 accessory protein BTK by ibrutinib. (Created with BioRender.com on 13 April 2022).
Similar results were also observed after anakinra treatment, which blocks the IL-1 receptor thus inhibiting downstream signalling
[20][21][22][78,88,89] (see
Figure 12).
The promising effect of anakinra treatment was also confirmed in a phase III clinical trial which showed significant reduction of COVID-19-related mortality when severe patients were treated with anakinra at early stages of the disease
[23][90].
The observation of NLRP3 inflammasome activation in vivo in COVID-19 patients and subsequent studies treating severe patients with experimental anti-pro-inflammatory cytokines allowed for the underlying molecular mechanisms to also be studied.
First of all, a landmark study conducted RNAseq on lung tissues from COVID-19-affected humans and compared the transcriptome with that of healthy lung donors. It was shown that NLRP3 signalling is upregulated in COVID-19 lungs, together with many ‘DAMPs’ such as metabolic dysregulation and ROS that are triggered by SARS-CoV-2 and can lead to NLRP3-inflammasome-mediated pyroptosis leading to a cytokine storm
[24][91] . After this significant indication that SARS-CoV-2 promoted NLRP3 activation
[24][91], besides the generic infection-associated signals, specific SARS-CoV-2 viral particle-mediated NLRP3 inflammasome activation has also been described on multiple levels.
Some knowledge derived from studies with SARS-CoV could be taken as indicative for the connection between SARS-CoV-2 and NLRP3. For example, it had been shown that SARS-CoV Orf3a could directly bind TRAF3 as well as ASC and activate the NLRP3 inflammasome
[25][92] (see
Figure 12).
Orf3a was able to induce both priming and activation signals of the NLRP3 inflammasome and lead to pyroptosis and mature IL-1β release. Although Orf3a belongs to the Viriporin pore-forming SARS-CoV protein family, SARS-CoV Orf3a-triggered NLRP3-mediated pyroptosis seemed to be independent of its pore-forming activity
[26][93]. The role of Orf3a in NLRP3 inflammasome activation was also investigated in the case of SARS-CoV-2. Interestingly, SARS-CoV-2 Orf3a was also found to induce NLRP3 inflammasome activation. However, this was based on the pore-forming activity of Orf3a
[27][94]. The discrepancies between the two mechanisms need to be further explored (see
Figure 12).
The Orf8b protein of SARS-CoV-2 was shown to be deadly on multiple levels. In non-myeloid cells that lack the NLRP3 inflammasome machinery, Orf8b accumulates and leads to ER stress, lysosomal damage, and caspase-independent cell death. However, in myeloid cells carrying NLRP3, it directly interacts with the NLRP3 LRR domain leading to ASC-speck formation and pyroptosis
[28][95]. Furthermore, it was found that the nucleocapsid of SARS-CoV-2 also binds directly to NLRP3 and leads to inflammasome activation and pyroptosis
[29][96] (see
Figure 12).
The non-structural protein NSP6 was also found to be a strong NLRP3 and pyroptosis initiator. NSP6 was shown to inhibit the lysosome–autophagosome system of the cell, which is known to trigger NLRP3 inflammasome formation
[30][97] (See
Figure 21).
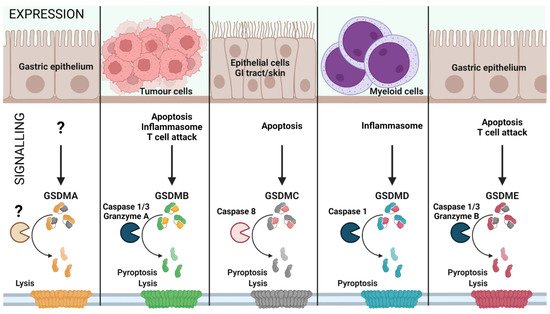
Figure 21. Gasdermin family: tissue-specific signalling. Gasdermin A/B/C/D/E are expressed in distinct cell types and are activated by various signals, leading to inflammatory or non-inflammatory cell lysis. The upstream events of GSDMA cleavage are not well characterized (represented as ‘?’). GSDMA downregulation in gastric epithelial cells can lead to tumour formation. Proliferating tumour cells are recognized by effector T cells that release Granzyme A, which cleaves GSDMB in cancer cells leading to pore formation and tumour cell lysis. Furthermore, GSDMB can also be activated by Caspase 1 or 3 downstream of inflammasome formation or apoptosis, leading to pyroptosis. GSDMC is activated by Caspase 8 downstream of apoptosis, linking apoptosis to pyroptosis. GSDMD is the best characterized GSDM effector molecule, cleaved downstream of inflammasome activation, leading to pyroptosis. Similar to GSDMC, GSDME links apoptosis to pyroptosis subsequent to cleavage by Caspase 3 in apoptotic cells. (Created with BioRender.com on 7 May 2022).
Orf3a was also associated with inhibition of autophagy by VPS39 interaction, highlighting another pathway of NLRP3 activation by Orf3a, in addition to pore formation and potassium efflux (see Figure 12).
However, the NLRP3 assembly trigger signal is not only associated with SARS-CoV-2. It was recently shown that the envelope protein of SARS-CoV-2 is a TLR2 ligand, and TLR2-SARS-CoV-2 envelope protein engagement leads to effective NLRP3 inflammasome priming. Blocking TLR2 during coronavirus infection thus leads to reduced inflammation
[31][98] (see
Figure 12).
SARS-CoV-2 infection manifests in neurological symptoms as well, which also seems to precipitate Parkinson´s disease
[32][99] according to a preprint publication that suggests that the SARS-CoV-2 spike protein activates the NLRP3 inflammasome ex vivo via ACE-2 interaction in human microglia. Additionally, a-synuclein, which is the underlying aggregate causing Parkinson´s disease, significantly enhanced inflammasome activation, which was completely dependent on NLRP3. The NLRP3-mediated pyroptosis in microglia might explain COVID-19-driven neuroinflammation
[33][100]. This
res
earchtudy, however, was not the first one to connect NLRP3 inflammasome priming and the SARS-CoV-2 spike protein. It was shown that the spike protein primes the NLRP3 inflammasome and leads to pyroptosis and IL-1β secretion ex vivo in cells derived from SARS-CoV-2-experienced individuals although not from healthy donors
[34][101]. Accordingly, SARS-CoV-2 infection renders macrophages in a distinct proinflammatory state that makes the spike protein even more immunogenic towards these immune cells. Interestingly, a different study showed that healthy monocytes do not express the ACE-2 receptor, while SARS-CoV-2-experienced monocytes express it in low levels, highlighting tissue-specific differences and SARS-CoV-2-mediated NLRP3 inflammasome activating pathways. It might very well be possible that the ACE-2 expression of SARS-CoV-2-experienced monocytes enables spike-specific NLRP3 inflammasome priming that was observed in the above
res
earchtudy [35][102]. SARS-CoV-2 infection of monocytes and macrophages is not necessarily dependent on spike-ACE-2 interaction. It can be Fc-receptor or complement-receptor mediated, or via antibody and complement opsonized virus
[36][37][103,104] (see
Figure 12).
Studying the SARS-CoV-2 genome itself provided further evidence that SARS-CoV-2 infection indeed leads to NLRP3 inflammasome activation: a cDNA screen of 28 SARS-CoV-2 ORFs revealed that the proteins NS1 and NS13 inhibit NLRP3 inflammasome activation and pyroptosis-mediated IL-1β release—a function which the virus would not invest in unless it posed a threat to its infectivity
[38][105].
As accumulating evidence supports the role of NLRP3 in the development of SARS-CoV-2-mediated inflammatory condition, targeting NLRP3 directly instead of the pyroptosis-related proinflammatory cytokines is emerging as a therapeutic strategy. Colchicine is an NLRP3 inhibiting molecule, although the exact underlying mechanism is not fully understood
[39][106]. Regardless, several clinical trials have been conducted to evaluate the beneficial effect of colchicine treatment in early SARS-CoV-2 infection
[40][107].
As the condition “long COVID-19” is also associated with a prolonged inflammatory state, an extended NLRP3 treatment period should also be considered.
Furthermore, after the identification of an FDA-approved inhibitor of the NLRP3-specific regulatory protein Bruton´s tyrosine kinase (BTK), the application of the commercially available BTK inhibitor ibrutinib in severe COVID-19 cases was investigated. First of all, a study showed that Waldenström Macroglobulinemia patients (a form of BTK-mediated B cell malignancy) that received ibrutinib cancer treatment had beneficial outcomes when contracting COVID-19, especially patients on high-dose ibrutinib treatment
[41][108]. A second study used off-label acalabrutinib (an ibrutinib-like BTK inhibitor) in severe COVID-19 patients which significantly increased the lung function of the patients
[42][109]. Although a direct link between NLRP3-mediated pyroptosis and ibrutinib treatment has not been established yet, the promising results call for further investigation of a broader ibrutinib treatment of COVID-19 patients (see
Figure 12).
Although increasing evidence supports the role of SARS-CoV-2-induced NLRP3 activation and pyroptosis in COVID-19 disease manifestation, one study showed that NLRP3 depletion or impaired pyroptosis had a negative effect on murine coronavirus infection outcomes in mice
[43][110].
However, as this researchtudy was conducted with a murine coronavirus MHV (mouse hepatitis virus), a direct comparison to human SARS-CoV-2 may be difficult.