Immune response has been shown to play an important role in defining patient prognosis and response to cancer treatment. Tumor-induced immunosuppression encouraged the recent development of new chemotherapeutic agents that assists in the augmentation of immune responses. Molecular mechanisms that tumors use to evade immunosurveillance are attributed to their ability to alter antigen processing/presentation pathways and the tumor microenvironment. Cancer cells take advantage of normal molecular and immunoregulatory machinery to survive and thrive. Cancer cells constantly adjust their genetic makeup using several mechanisms such as nucleotide excision repair as well as microsatellite and chromosomal instability, thus giving rise to new variants with reduced immunogenicity and the ability to continue to grow without restrictions.
- immunosuppression
- Cancer
- Signaling Pathways
1. Introduction
2. Immune Evasion
Cancer cells are able to hijack the immune system by secreting cytokines and molecules familiar to effector T cells enabling them to evade immunosurveillance. Immunosurveillance is a process whereby the immune system guards against and averts cancer progression. Immunosurveillance is a concept that was first hypothesized by Paul Ehrlich[10] in 1909 when he proposed that the immune system restricted the growth of carcinomas. Five decades later, Burnet FA [11] and Thomas L [12] presented theories that supported Ehrlich’s theory. Burnet suggested that there might be a tumor-specific immune response that attempts to destroy developing cancer, whilst Thomas thought that there must be a mechanism similar to that of the host immune system versus foreign tissue, commonly seen in graft rejection, in which cancer can be fought off by the immune response. Even though this theory was proven correct, cancer cells still had a way of progressing, and that is when the concept of tumor immunoediting was hypothesized. This concept is divided into three different phases: elimination, equilibrium, and escape [13][14].2.1. Elimination
The antitumor immune response is initiated by the activation of the innate immune system in the presence of cancer cells. Cells of the innate immune system are altered to favor proangiogenic activities and an immunosuppressive microenvironment. One of the major angiogenic factors, vascular endothelial growth factor (VEGF), limits tumor-infiltrating T cells and antigen-presenting cells (APCs) activity to foster immunosuppressive microenvironment through upregulation of Tregs and immune checkpoint inhibitors [15]. Exposure to carcinogens such as tobacco smoke or asbestos has been correlated with tissue disruption/inflammation through the activation of IL-1β, which enhances their tumorigenic ability [16]. Cancer cells stress promotes the production of other proinflammatory cytokines and proteins such as heat-shock proteins (HSPs) [17] and Natural killer group 2, member D (NKG2D), which serve as danger signals. Natural killer (NK) cells, macrophages, γδ T cells, and NK T cells are released to the tumor site resulting in cytotoxic effector mechanisms to eliminate cancer cells [18].2.2. Equilibrium
Equilibrium refers to a period between the failure of the immune system to completely eliminate/eradicate cancer cells and the beginning of the escape phase. This is the period where the malignant disease is clinically detectable. Cancer cells constantly adjust their genetic makeup either via nucleotide excision repair, microsatellite instability, or chromosomal instability, giving rise to new phenotypes that display reduced immunogenicity [19][20][21]. These cancer cells evolve with the generation of more advanced mutations that provide increased resistance to immunological attack until both immune and cancer cells are at an equilibrium state. These new variants of cancer cells have the ability to progress to the escape phase of the immunoediting process [22]. Furthermore, cancer cells induce alterations in the genome-processing mechanisms such as deoxyribonucleic acid (DNA) damage–repair machinery, telomere damage, centrosome amplification, and epigenetic modifications to develop new variants [23][24]. This is accomplished by hijacking processes that are mainly involved in cell division and tumor suppression [25]. Defective DNA damage repair results in the accumulation of immunosuppressive cells and decreased T cell responses [26]. These alterations have also been shown to contribute immensely to immunotherapeutic responses in cancer [27] and molecularly targeted cancer therapies. Of note, there are several key molecular signaling pathways identified that are associated with cancer progression and drug resistance. The most common of these pathways is the phosphatidylinositol 3-kinase (PI3K)/AKT/mTOR (tumor survival pathway) and its interrelated pathways, which will be discussed in more detail later in this review. The mechanism of action by inhibitors of this pathway includes the induction of DNA damage, particularly in cancers that take advantage of the DNA damage–repair system. Hence, dysfunctional production of nucleotides necessary for DNA synthesis and repair are the main components that allow for treatment efficacy with PI3K inhibitors (Figure 1).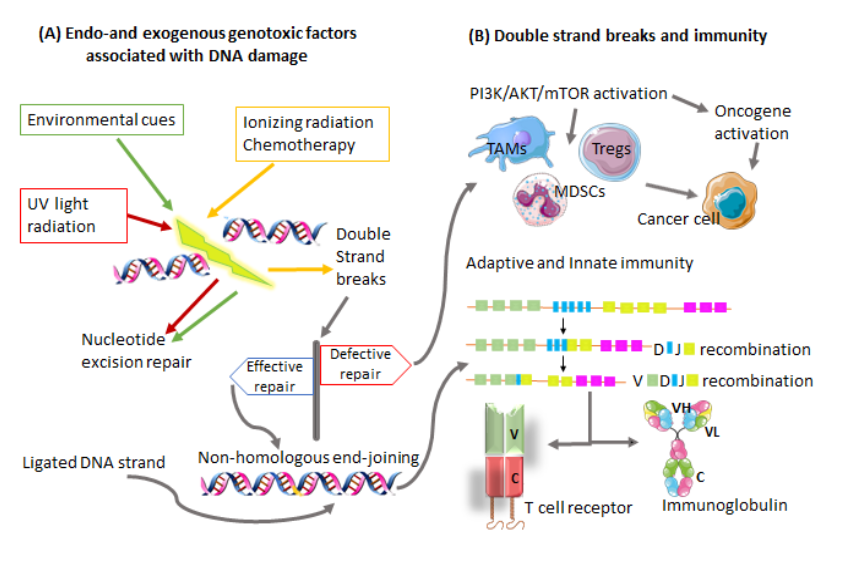 Figure 1: Impaired transcription and replication process can occur, leading to DNA damage and genomic instability. (A) DNA damage can be a result of multiple factors, which could be of an endo or exogenous nature. (B) Repair of double-strand breaks (DSBs) is crucial to cell development and survival. DSBs are repaired without the need for a homologous template by the nonhomologous end-joining pathway. Consequently an efficiently ligated DNA strand results in a heterogenous pool of antigen receptor genes needed for T and B cell development. Failure of the DSB repair mechanism activates the PI3K/AKT/mTOR signaling pathway, which induces immunosuppressive cells known to promote cancer progression. Variable region (v), Constant region (c), Heavy chain (H), Light chain (L).
2.3. Escape
Cancer cells with an altered genetic makeup have the ability to withstand the immunological stress throughout the equilibrium stage and proceed to the escape phase, where they continue to grow without restrictions. The mechanisms utilized by cancer cells to proceed to the escape phase are attributed to their ability to alter antigen processing and presentation pathways.
Figure 1: Impaired transcription and replication process can occur, leading to DNA damage and genomic instability. (A) DNA damage can be a result of multiple factors, which could be of an endo or exogenous nature. (B) Repair of double-strand breaks (DSBs) is crucial to cell development and survival. DSBs are repaired without the need for a homologous template by the nonhomologous end-joining pathway. Consequently an efficiently ligated DNA strand results in a heterogenous pool of antigen receptor genes needed for T and B cell development. Failure of the DSB repair mechanism activates the PI3K/AKT/mTOR signaling pathway, which induces immunosuppressive cells known to promote cancer progression. Variable region (v), Constant region (c), Heavy chain (H), Light chain (L).
2.3. Escape
Cancer cells with an altered genetic makeup have the ability to withstand the immunological stress throughout the equilibrium stage and proceed to the escape phase, where they continue to grow without restrictions. The mechanisms utilized by cancer cells to proceed to the escape phase are attributed to their ability to alter antigen processing and presentation pathways.
2.3.1. Escaping the Antigen Presentation Pathway
In a normal setting, tumor-associated antigens will be presented to cytotoxic T cells through major histocompatibility complex (MHC) class I; however, cancer cells downregulate the expression of proteins involved in antigen presentation including MHC I proteins and inhibit the maturation of DCs. This dispossesses cytotoxic T cells’ ability to recognize tumor cells, thus allowing them to evade immunosurveillance [28]. Downregulation or loss of MHC class I molecule expression could result from the heterogeneous expression of multiple tumor antigens that develop due to mutations in the β2 macroglobulin subunit [29][30]. Activated APCs destroy cancer cells by either engulfing them or through interaction with tumor-infiltrating NK cells. The NK cells’ method of destroying cancer cells can still be utilized to destroy cells with downregulated expression of MHC class I molecules [28]. To escape this, cancer cells develop other mechanisms such as downregulation of low molecular mass polypeptide (LMP) 2 and 3, which results in modifications of various antigens presented by MHC class I molecules [31].2.3.2. The PD-1/PD-L1 Pathway as a Mechanism of Escape
Programmed cell death protein-1 (PD-1) and its ligand PD-L1 can both be expressed on the surfaces of cancer cells, whilst PD-1 is predominantly expressed on the surface of immune cells. The PD-L1/PD-1 pathway’s primary function is the maintenance of immune tolerance and protecting the body from self-harm through the immunological attack. The downside of PD-L1/PD-1 pathways is that hindering T cells’ immune responses also provides a way for cancer cells to evade the immune system and survive. The immunosuppressive ability of this pathway encouraged the development of inhibitors against PD-L1/PD-1 proteins as cancer immunotherapy [32][33].3. The Tumor Microenvironment
The tumor microenvironment consists of a diverse population of nonmalignant cells/components, including immune cells, fibroblasts, stem cells, endothelial cells, secreted proteins, extracellular matrix, and blood vessels that can be manipulated by cancer cells to promote its proliferation and survival. Tumor cells accomplish this by establishing harmonious cross talk and interaction with the components of the tumor microenvironment. The components of the tumor microenvironment and the structure can differ according to cancer types [34][35]. Cells of the tumor microenvironment, particularly T lymphocytes, have been used in adoptive cell therapies either autologously or from allogeneic donors. Their use as a clinical diagnostic tool is well established, with more studies venturing on finding related biomarkers that can be used as predictors of patient clinical outcomes. Activation of hypoxia-inducible factor (HIF) signaling promotes cellular adaptation to hypoxic conditions. This gives cancer cells a unique phenotype that allows them to survive and grow beyond control with the ability to resist cancer therapy [36]. As one of the key factors that promote cancer progression, HIF regulates several processes within the tumor microenvironment. The mTOR pathway significantly induces HIF-1α. However, reduction in nutrition supply in cancer cells inhibits HIF-1α activity via a mTORC1 dependent mechanism [37]. The HIF-1/2α upregulates genes that assume control of immunomodulatory and metabolic processes within the tumor microenvironment. A process accomplished by induction of epithelial–mesenchymal transition (EMT)-related transcription factors such as SNAIL and ZEB family of transcription factors, and twist transcription factor (TWIST), to mention a few [38]. The EMT processes have also been implicated in cancer progression, metastasis, and drug resistance [39]. Of interest, the mTOR signaling pathway is associated with numerous immunosuppressive cells that contribute substantially to the development of a suitable tumor microenvironment for cancer progression and drug resistance. High mTORC1 activity and increased glycolytic metabolism were observed in effector Tregs similar to that seen in CD8+ T cells. Effector Tregs also showed higher activity of HIF-α and glycolysis enzymes, hexokinase 2 (Hk2), and phosphofructose kinase (Pfkp). Furthermore, effector Tregs mTORC1 activity was not comparable to their central Tregs counterparts during the analysis of glycolysis and TCA cycle metabolites. Overall, this data suggests that antigen exposed Tregs have high mTOR and glycolysis [40].4. Immunoregulatory Signaling Pathways
4.1. Myeloid-Derived Suppressor Cells
The MDSCs are a heterogeneous population of immature immune cells derived from a common myeloid progenitor within the bone marrow [41]. These cells will subsequently be differentiated into monocytes, macrophages, DCs, and granulocytes [42]. Their immunosuppressive effect is associated with the worst patient prognosis in cancer [43]. High levels of circulating MDSCs were also associated with the worst overall survival in patients with solid tumors suggesting the importance of these cells as potential therapeutic targets for the treatment of the disease [44]. They have also been implicated in reducing treatment response to immune checkpoint inhibitors [8]. Myeloid-related protein S100A9 has been shown to be one of the mechanisms that cancer cells use to block antitumor mechanisms. Overexpression of S100A9 increases the levels of MDSCs, which are associated with impaired maturation of APCs within the tumor microenvironment [45]. Multiple proteins are also involved in the regulation of MDSCs in cancer. Expressed mainly by hemopoietic cells and encoded by the inpp5d gene [46], SHIP-1 is a negative regulator of the PI3K/AKT downstream signaling pathway in a number of cellular activation processes, including myeloid survival. SHIP expression has been shown to be essential in the maintenance of myeloid cells. The downregulation of SHIP expression induced apoptosis in neutrophils and mast cells whilst downstream regulation of PI3K by AKT is associated with reduced apoptosis [47][48][49]. Neutrophils have been shown to promote cancer progress via degradation of insulin receptor substrate-1 (IRS-1). This result promotes PI3K interaction with mitogen platelet-derived growth factor receptor (PDGFR and resultant tumorigenesis [49]. In the absence of SHIP, AKT becomes phosphorylated, an effect abrogated by inhibition of PI3K, indicating its role as the key regulator of AKT [48]. The function of SHIP and its multiple immunomodulation of signaling pathways is discussed in more detail elsewhere [50]. The induction of PI3K activation is increased in myeloproliferative diseases such as acute myeloid leukemia [51]. Efforts to block PI3K along with mTOR signaling pathways for the implementation of molecularly targeted therapies in clinical trials are ongoing [28]. The parallel interaction between mTOR and PI3K signaling pathways implies their similarity in performing regulatory roles in cell proliferation and apoptosis needed for cancer survival and proliferation [29]. Thus, the use of SHIP alone or in combination with inhibitors of PI3K and parallel pathways in cancer has been postulated. One of the known signaling pathways that runs parallel with PI3K is mitogen-activated protein kinases (MAPK) signaling.4.2. Tumor-Associated Macrophages
Macrophages that infiltrate solid tumor microenvironments are referred to as tumor-associated macrophages (TAMs) [30]. These macrophages have been found in abundance in a number of cancers, including breast [31], colorectal [32], pancreatic [33], and prostate [34] cancers. High infiltration of macrophages within the tumor microenvironment is associated with reduced overall survival and treatment response. TAMs were found to be mainly of the M2 macrophage lineage, and their increased production of anti-inflammatory factors contributes to tumor progression [35]. The balance between TAMs and M1 macrophages is determined by signaling pathways such as the STAT pathway. The M1 macrophage polarizing signals induced by IFN-γ and lipopolysaccharide (LPS) activate the STAT1 pathway whilst STAT3/6 pathways are activated by M2 macrophage polarizing cytokines such as IL-10, IL-4, and IL-13 [36]. To tilt the scale towards the M2 phenotype, which assists in cancer progression, the kruepper-like 2 (KFL2) transcription factor, along with STAT6, induces M2 genes Arg-1, Mrc1, Fizz1, and PPARγ. In the same manner, M1 genes TNF-α, Cox-2, CCL5, and iNOS are blocked via the NF-κB/Hypoxia-inducible factor 1-alpha (HIF-1α) pathway [37]. Contradictory to these findings, STAT6 driven inhibition of the M2 polarization is achieved by Trim24 CREB-binding protein (CBP)-associated E3 ligase acetylation [38]. The activated STAT6 pathway can also induce the M2 phenotype via the IL-4 pathway, which is associated with lung cancer progression and is inactive in the M1 phenotype [39]. The switch from M1 to M2 macrophages is mediated by IRF/STAT signaling [40], while the LPS stimulated TLR4 will switch polarization towards the M1 phenotype. Thus, both the NF-κB/HIF-1α and IRF/TLR/STAT signaling pathways could be targeted in cancer to prevent cancer cells from persistently shifting macrophages into TAMs, which favor cancer progression within the tumor microenvironment. The PI3K/AKT signaling pathway seems to play a crucial role in immunoregulatory cells, including TAMs. By coculturing TAMs with lung adenocarcinoma cells, higher expression levels of PI3K/AKT proteins were observed [41]. The PI3K pathway has also been shown to be highly involved in macrophage polarization. The absence of PI3Kγ is associated with the M1 polarization [42], and activation of PI3K leads to M2 polarization [43]. The Hedgehog signaling pathway has been shown to play a role in cancer progression [44]. Cancer cells secrete sonic Hedgehog (SHH) to promote their proliferation and survival. To do so, Hedgehog facilitates macrophage polarization within the tumor microenvironment into the protumor M2 phenotype [45]. Aberrant signaling of the Hedgehog pathway is associated with dysregulated tissue patterning and development, leading to a number of pediatric cancers.4.3. Regulatory T Cells
Regulatory T cells (Tregs) are known as one of the “master” immunoregulatory cells designed to maintain immune homeostasis. However, cancer cells take advantage of their suppressive effect on T cells to evade the immune response and continue to grow without restriction. Tregs are a heterogeneous set of immune cells. This means that finding a specific marker, particularly in humans, for inhibition strategies remains a challenge. Efforts to boost anticancer immune responses by blocking suppressive Tregs mechanisms are still being explored. This includes inhibition of Tregs-related suppressive cytokines and surface markers using antibodies [46]. For instance, the expression of the inhibitory IL-35 cytokine and chemokine receptors such as CCR5 recruits Tregs and activates AKT/mTOR signaling pathway to promote Tregs function [47]. The suppressive function of Tregs is known to be a contributing factor to cancer progression. The AKT/mTOR signaling pathway, generally known to be activated in cancer, is also responsible for cancer progression. Therapeutic interventions aimed at targeting these pathways have been implemented, with some being in clinical trials with positive patient responses [48]. Upon further investigation, cancer-related signaling pathways, P53 hypoxia, TNF receptor-associated factor 6–mediated (TRAF6-mediated), IFN regulatory factor 7 (IRF-7) activation, NKT pathway and inhibitory immune checkpoint receptors, T cell immunoreceptor with immunoglobulin and ITIM domains (TIGIT), PD-1, and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) were found to be upregulated in mice splenic Tregs [47].5. Personalized Precision Medicine and Combinatorial Therapies
Molecular testing platforms are employed to detect abnormalities between normal/cancerous tissue and the blood. Alterations in DNA, RNA, splicing factors, and post-translational modifications are used for diagnostic purposes, prediction of possible future development of disease mainly due to inheritance, and the development of therapies [49]. Molecularly targeted therapies are aimed at targeting these aberrant signatures either by down or upregulation of the genes relevant to a particular disease. Personalized precision medicine is aimed at developing individualized therapeutic strategies that are more effective at treating a specific type of disease with fewer adverse events and reduced therapeutic resistance [50]. Even though there is less toxicity and reduced adverse events compared with chemotherapy, targeted therapies have presented their own drawbacks in this regard. A recent study by Du et al. picked up a high incidence of adverse events in targeted therapies. The top five on their list included skin damage, fatigue, mucosal damage, hypertension, and gastrointestinal discomfort. A combination of molecularly targeted therapies with existing or newly developed chemoimmunotherapies should therefore be considered. All three immunosuppressive cells (MDSCs, TAMs, and Tregs) have a dysregulated PI3K signaling pathway as a common factor. Therapeutic interventions targeting PI3K are available, with some still in clinical trials (Table 1). However, the efficacy of PI3K inhibitors is limited by therapeutic resistance. Some of the methods suggested to overcome drug resistance are a reactivation of the PI3K signaling pathway in combination with parallel pathways such as (but not limited to) the AKT/mTOR signaling network and manipulation of the tumor microenvironment [46]. However, it was shown that the response rate can still be very low in some cases, such as treatment of triple-negative breast cancer patients with alterations in PIK3CA/AKT1/PTEN using buparlisib. Only three out of six patients with targeted DNA sequencing (MSK-IMPACT) had stable disease indicating the ineffectiveness of buparlisib in treatment in this cohort of patients [51].6. Conclusions and Future Perspectives
Most cancer therapeutic endeavors are aimed at targeting a particular molecule at a time, but medicine is slowly revolutionizing, and multiple sets of proteins in combination with key molecular signaling pathways are being targeted for the development of more directed/personalized therapies. More and more studies are exploring the use of less invasive methods to diagnose or direct treatment decisions in cancer, and circulating immune cells have been at the forefront of these endeavors. Of note, SHIP, SHH, and SECs in MDSCs, TAMs, and Tregs, respectively, can be targeted in combination with the PI3K signaling pathway and related molecules in cancers where these cells serve as one of the prominent biomarkers of the disease or are associated with poor clinical outcome. In other instances where immunosuppressive cells serve as indicators of drug resistance, particularly in relation to immune checkpoint inhibitors, personalized combinatorial therapies can be explored to decipher drug resistance, including cases where treatment with PI3K inhibitors is ineffective. Realization of curative cancer strategies can be accomplished through tackling both immunological and molecular signaling pathways. To do so, researchers from different facets of cancer research can develop the ultimate cancer munition by combining specialties, as in the case of the use of immunotherapy or nanoparticle to enhance radiotherapeutic responses. Patient stratification can be performed through immunological and molecular biomarkers. This will assist in selecting patients who will better tolerate certain types of immunological treatment whilst exposing them to molecularly targeted therapies as well. An attempt to boost anticancer immunity while blocking the function of immunosuppressive cells can be achieved by blocking related molecular signaling pathways, which have also been shown to be activated in most cancers.| PI3K Inhibitor | Mode of Action | Cancer Type | References |
|---|---|---|---|
| Alpelisib | PIK3CA/PI3K-δ isoform | Hormone receptor +/HER2-Breast Cancer | [52][53] |
| Copanlisib | PI3Kβ, PI3Kγ, PI3K-α & PI3K-δ isoforms | Follicular Lymphoma | [54] |
| Duvelisib | PI3K-δ and PI3K γ isoforms | Chronic Lymphocytic Leukemia | [55] |
| Idelalisib | PI3Kδ | Chronic Lymphocytic Leukemia | [56] |
| Buparlisib | PI3Kα, PI3Kβ, PI3Kδ and PI3Kγ | Metastatic triple-negative breast and colorectal cancers | [51][57] |
| Pictilisib | PI3Kα and PI3Kδ | Advanced breast cancer and cancer of the bone | [58][59] |
References
- Li Kai, Luo Haiqing, Huang Lianfang, Luo Hui, Zhu Xiao; Microsatellite instability: A review of what the oncologist should know. Cancer Cell Int 2020, 20, 16, 10.1186/s12935-019-1091-8. .
- do Canto, L. M; Larsen, S. J; Catin Kupper, B. E; Begnami, M; Scapulatempo-Neto, C; Petersen, A. H; Aagaard, M. M; Baumbach, J; Aguiar, S Jr; & Rogatto, S. R.; et al. Increased Levels of Genomic Instability and Mutations in Homologous Recombination Genes in Locally Advanced Rectal Carcinomas. Frontiers in oncology 2019, 9, 395, 10.3389/fonc.2019.00395.
- Giwon Shin; Stephanie U. Greer; Erik Hopmans; Susan M. Grimes; Hojoon Lee; Lan Zhao; Laura Miotke; Carlos Suarez; Alison F. Almeda; Sigurdis Haraldsdottir; et al.Hanlee P. Ji Profiling diverse sequence tandem repeats in colorectal cancer reveals co-occurrence of microsatellite and chromosomal instability involving Chromosome 8. Genome Medicine 2021, 13, 1-18, 10.1186/s13073-021-00958-z.
- Jardim J. Melanie; Wang Qinhong; Furumai Ryohei; Wakeman Timothy; Goodman K. Barbara; Wang Xiao-Fan; Reduced ATR or Chk1 Expression Leads to Chromosome Instability and Chemosensitization of Mismatch Repair–deficient Colorectal Cancer Cells. Mol. Biol. Cell 2009, 20, 3801–3809, doi.org/10.1091/mbc.e09-04-0303.
- Chen Mengting; Renske Linstra; van Vugt A.T.M. Marcel; Genomic instability, inflammatory signaling and response to cancer immunotherapy. Biochim. et Biophys. Acta (BBA) Rev. Cancer 2022, 1877, 188661, https://doi.org/10.1016/j.bbcan.2021.188661.
- Ryungsa Kim; Manabu Emi; Kazuaki Tanabe; Koji Arihiro; Tumor-Driven Evolution of Immunosuppressive Networks during Malignant Progression. Cancer Research 2006, 66, 5527-5536, 10.1158/0008-5472.can-05-4128.
- DaQian Gu; Xiang Ao; Yu Yang; Zhuo Chen; Xiang Xu; Soluble immune checkpoints in cancer: production, function and biological significance. Journal for ImmunoTherapy of Cancer 2018, 6, 132, 10.1186/s40425-018-0449-0.
- Rebekka Weber; Viktor Fleming; Xiaoying Hu; Vasyl Nagibin; Christopher Groth; Peter Altevogt; Jochen Utikal; Viktor Umansky; Myeloid-Derived Suppressor Cells Hinder the Anti-Cancer Activity of Immune Checkpoint Inhibitors. Frontiers in Immunology 2018, 9, 1310, 10.3389/fimmu.2018.01310.
- Ruth Nussinov; Chung-Jung Tsai; Hyunbum Jang; A New View of Pathway-Driven Drug Resistance in Tumor Proliferation. Trends in Pharmacological Sciences 2017, 38, 427-437, 10.1016/j.tips.2017.02.001.
- Ehrlich, P; Ueber den jetzigen stand der karzinomforschung. vortrag gehalten vor den studenten der amsterdamer universitaet, vereinigung fuer wissenschaftliche arbeit 1 june 1908.. Beitraege Exp. Pathol. Chemother. Akad. Verl. Leipz 1909, 118, 164.
- MacFarlane Burnet; Cancer--A Biological Approach: III. Viruses Associated with Neoplastic Conditions. IV. Practical Applications. BMJ 1957, 1, 841-847, 10.1136/bmj.1.5023.841.
- L Thomas; On immunosurveillance in human cancer.. The Yale journal of biology and medicine 1982, 55, 329-33.
- Gavin P. Dunn; Lloyd J. Old; Robert D. Schreiber; The Immunobiology of Cancer Immunosurveillance and Immunoediting. Immunity 2004, 21, 137-148, 10.1016/j.immuni.2004.07.017.
- Joanina K. Gicobi; Whitney Barham; Haidong Dong; Immune resilience in response to cancer therapy. Cancer Immunology, Immunotherapy 2020, 69, 2165-2167, 10.1007/s00262-020-02731-4.
- Adriana Albini; Antonino Bruno; Douglas M. Noonan; Lorenzo Mortara; Contribution to Tumor Angiogenesis From Innate Immune Cells Within the Tumor Microenvironment: Implications for Immunotherapy. Frontiers in Immunology 2018, 9, 527, 10.3389/fimmu.2018.00527.
- Sergei I. Grivennikov; Florian Greten; Michael Karin; Immunity, Inflammation, and Cancer. Cell 2010, 140, 883-899, 10.1016/j.cell.2010.01.025.
- Mark G Goldstein; Zihai Li; Heat-shock proteins in infection-mediated inflammation-induced tumorigenesis. Journal of Hematology & Oncology 2009, 2, 5-5, 10.1186/1756-8722-2-5.
- I. Seren Bernardone; Role of NK cells and adaptive immunity in “immunoediting”: Recent developments. Inmunología 2008, 27, 141-146, 10.1016/s0213-9626(08)70062-3.
- Jing Zhang; David J. H. Shih; Shiaw-Yih Lin; Role of DNA repair defects in predicting immunotherapy response. Biomarker Research 2020, 8, 1-8, 10.1186/s40364-020-00202-7.
- Myrthe Jager; Francis Blokzijl; Ewart Kuijk; Johanna Bertl; Maria Vougioukalaki; Roel Janssen; Nicolle Besselink; Sander Boymans; Joep de Ligt; Jakob Skou Pedersen; et al.Jan HoeijmakersJoris PothofRuben van BoxtelEdwin Cuppen Deficiency of nucleotide excision repair is associated with mutational signature observed in cancer. Genome Research 2019, 29, 1067-1077, 10.1101/gr.246223.118.
- Natalia Vargas-Rondón; Victoria E. Villegas; Milena Rondón-Lagos; The Role of Chromosomal Instability in Cancer and Therapeutic Responses. Cancers 2017, 10, 4, 10.3390/cancers10010004.
- Ryungsa Kim; Manabu Emi; Kazuaki Tanabe; Cancer immunoediting from immune surveillance to immune escape. Immunology 2007, 121, 1-14, 10.1111/j.1365-2567.2007.02587.x.
- Lynnette R. Ferguson; Helen Chen; Andrew R. Collins; Marisa Connell; Giovanna Damia; Santanu Dasgupta; Meenakshi Malhotra; Alan K. Meeker; Amedeo Amedei; Amr Amin; et al.S. Salman AshrafKatia AquilanoAsfar S. AzmiDipita BhaktaAlan BilslandChandra S. BoosaniSophie ChenMaria Rosa CirioloHiromasa FujiiGunjan GuhaDorota HalickaWilliam G. HelferichW. Nicol KeithSulma I. MohammedElena NiccolaiXujuan YangKanya HonokiVirginia R. ParslowSatya PrakashSarallah RezazadehRodney E. ShackelfordDavid SidranskyPhuoc T. TranEddy S. YangChristopher A. Maxwell Genomic instability in human cancer: Molecular insights and opportunities for therapeutic attack and prevention through diet and nutrition. Seminars in Cancer Biology 2015, 35, S5-S24, 10.1016/j.semcancer.2015.03.005.
- Yixin Yao Wei Dai; Genomic Instability and Cancer. Journal of Carcinogenesis & Mutagenesis 2014, 5, 1000165, 10.4172/2157-2518.1000165.
- Yixin Yao Wei Dai; Genomic Instability and Cancer. Journal of Carcinogenesis & Mutagenesis 2014, 5, 1000165, 10.4172/2157-2518.1000165.
- Daan K. J. Pieren; Noortje A. M. Smits; Sandra Imholz; Bhawani Nagarajah; Conny T. van Oostrom; Renata M. C. Brandt; Wilbert P. Vermeij; Martijn E. T. Dollé; Teun Guichelaar; Compromised DNA Repair Promotes the Accumulation of Regulatory T Cells With an Aging-Related Phenotype and Responsiveness. Frontiers in Aging 2021, 2, 13, 10.3389/fragi.2021.667193.
- Laetitia Nebot-Bral; Clélia Coutzac; Patricia L. Kannouche; Nathalie Chaput; Why is immunotherapy effective (or not) in patients with MSI/MMRD tumors?. Bulletin du Cancer 2018, 106, 105-113, 10.1016/j.bulcan.2018.08.007.
- Salihanur Darici; Hazem AlKhaldi; Gillian Horne; Heather G. Jørgensen; Sandra Marmiroli; Xu Huang; Targeting PI3K/Akt/mTOR in AML: Rationale and Clinical Evidence. Journal of Clinical Medicine 2020, 9, 2934, 10.3390/jcm9092934.
- Zhilin Zou; Tao Tao; Hongmei Li; Xiao Zhu; mTOR signaling pathway and mTOR inhibitors in cancer: progress and challenges. Cell & Bioscience 2020, 10, 1-11, 10.1186/s13578-020-00396-1.
- Luca Cassetta; Jeffrey W. Pollard; Tumor-associated macrophages. Current Biology 2020, 30, R246-R248, 10.1016/j.cub.2020.01.031.
- Amanda Valeta-Magara; Abhilash Gadi; Viviana Volta; Beth Walters; Rezina Arju; Shah Giashuddin; Hua Zhong; Robert J. Schneider; Inflammatory Breast Cancer Promotes Development of M2 Tumor-Associated Macrophages and Cancer Mesenchymal Cells through a Complex Chemokine Network. Cancer Research 2019, 79, 3360-3371, 10.1158/0008-5472.can-17-2158.
- Yahui Zhao; Weina Zhang; Miaomiao Huo; Peng Wang; Xianghe Liu; Yu Wang; Yinuo Li; Zhixiang Zhou; Ningzhi Xu; Hongxia Zhu; et al. XBP1 regulates the protumoral function of tumor-associated macrophages in human colorectal cancer. Signal Transduction and Targeted Therapy 2021, 6, 1-14, 10.1038/s41392-021-00761-7.
- Samantha B Kemp; Nina G Steele; Eileen S Carpenter; Katelyn L Donahue; Grace G Bushnell; Aaron H Morris; Stephanie The; Sophia M Orbach; Veerin R Sirihorachai; Zeribe C Nwosu; et al.Carlos EspinozaFatima LimaKristee BrownAlexander A GirgisValerie GunchickYaqing ZhangCostas A LyssiotisTimothy L FrankelFilip BednarArvind RaoVaibhav SahaiLonnie D SheaHoward C CrawfordMarina Pasca di Magliano Pancreatic cancer is marked by complement-high blood monocytes and tumor-associated macrophages. Life Science Alliance 2021, 4, e202000935, 10.26508/lsa.202000935.
- Prahara Yuri; Katsumi Shigemura; Koichi Kitagawa; Exsa Hadibrata; Muhammad Risan; Andy Zulfiqqar; Indrawarman Soeroharjo; Ahmad Z. Hendri; Raden Danarto; Aya Ishii; et al.Saya YamasakiYongmin YanDidik S. HeriyantoMasato Fujisawa Increased tumor-associated macrophages in the prostate cancer microenvironment predicted patients’ survival and responses to androgen deprivation therapies in Indonesian patients cohort. Prostate International 2020, 8, 62-69, 10.1016/j.prnil.2019.12.001.
- S. A. Almatroodi; C. F. McDonald; I. A. Darby; D. S. Pouniotis; Characterization of M1/M2 Tumour-Associated Macrophages (TAMs) and Th1/Th2 Cytokine Profiles in Patients with NSCLC. Cancer Microenvironment 2015, 9, 1-11, 10.1007/s12307-015-0174-x.
- Antonio Sica; Vincenzo Bronte; Altered macrophage differentiation and immune dysfunction in tumor development. Journal of Clinical Investigation 2007, 117, 1155-1166, 10.1172/jci31422.
- Antonio Sica; Alberto Mantovani; Macrophage plasticity and polarization: in vivo veritas. Journal of Clinical Investigation 2012, 122, 787-795, 10.1172/jci59643.
- Tao Yu; Shucheng Gan; Qingchen Zhu; Dongfang Dai; Ni Li; Hui Wang; Xiaosong Chen; Dan Hou; Yan Wang; Qiang Pan; et al.Jing XuXingli ZhangJunli LiuSiyu PeiChao PengPing WuSimona RomanoChaoming MaoMingzhu HuangXiaodong ZhuKunwei ShenJun QinYichuan Xiao Modulation of M2 macrophage polarization by the crosstalk between Stat6 and Trim24. Nature Communications 2019, 10, 1-15, 10.1038/s41467-019-12384-2.
- Cuiping Fu; Liyan Jiang; Shengyu Hao; Zilong Liu; Suling Ding; Weiwei Zhang; Xiangdong Yang; Shanqun Li; Activation of the IL-4/STAT6 Signaling Pathway Promotes Lung Cancer Progression by Increasing M2 Myeloid Cells. Frontiers in Immunology 2019, 10, 2638, 10.3389/fimmu.2019.02638.
- Nan Wang; Hongwei Liang; Ke Zen; Molecular Mechanisms That Influence the Macrophage M1–M2 Polarization Balance. Frontiers in Immunology 2014, 5, 614, 10.3389/fimmu.2014.00614.
- Shiyang Yuan; Yaling Dong; Laishui Peng; Mei Yang; Linxia Niu; Zhiwen Liu; Junping Xie; Tumor‑associated macrophages affect the biological behavior of lung adenocarcinoma A549 cells through the PI3K/AKT signaling pathway. Oncology Letters 2019, 18, 1840-1846, 10.3892/ol.2019.10483.
- Mariane T. Amano; Angela Castoldi; Vinicius Andrade-Oliveira; Marcela T. Latancia; Fernanda F. Terra; Matheus Correa-Costa; Cristiane N.S. Breda; Raphael J.F. Felizardo; Welbert O. Pereira; Marina B. da Silva; et al.Mariana Y.S. MiyagiCristhiane F. AguiarMeire I. HiyaneJoão S. SilvaIvan Cruz MouraNiels O.S. Camara The lack of PI3Kγ favors M1 macrophage polarization and does not prevent kidney diseases progression. International Immunopharmacology 2018, 64, 151-161, 10.1016/j.intimp.2018.08.020.
- Chunmei Liu; Bohui Li; Kaihong Tang; Xuening Dong; Longge Xue; Guangming Su; Yingyu Jin; Aquaporin 1 alleviates acute kidney injury via PI3K-mediated macrophage M2 polarization. Inflammation Research 2020, 69, 509-521, 10.1007/s00011-020-01334-0.
- Marina Pasca di Magliano; Matthias Hebrok; Hedgehog signalling in cancer formation and maintenance. Nature Reviews Cancer 2003, 3, 903-911, 10.1038/nrc1229.
- Amy J. Petty; Ang Li; Xinyi Wang; Rui Dai; Benjamin Heyman; David Hsu; XiaoPei Huang; Yiping Yang; Hedgehog signaling promotes tumor-associated macrophage polarization to suppress intratumoral CD8+ T cell recruitment. Journal of Clinical Investigation 2019, 129, 5151-5162, 10.1172/jci128644.
- Jing Yang; Ji Nie; Xuelei Ma; Yuquan Wei; Yong Peng; Xiawei Wei; Targeting PI3K in cancer: mechanisms and advances in clinical trials. Molecular Cancer 2019, 18, 1-28, 10.1186/s12943-019-0954-x.
- Ying Shao; William Y. Yang; Fatma Saaoud; Charles Drummer; Yu Sun; Keman Xu; Yifan Lu; Huimin Shan; Ethan M. Shevach; Xiaohua Jiang; et al.Hong WangXiaofeng Yang IL-35 promotes CD4+Foxp3+ Tregs and inhibits atherosclerosis via maintaining CCR5-amplified Treg-suppressive mechanisms. JCI Insight 2021, 6, e152511, 10.1172/jci.insight.152511.
- Ali S. Alzahrani; PI3K/Akt/mTOR inhibitors in cancer: At the bench and bedside. Seminars in Cancer Biology 2019, 59, 125-132, 10.1016/j.semcancer.2019.07.009.
- Nakamura, R.M.; Kasahara, Y. Chapter 19—Molecular Diagnostics in the Evaluation of Cancer: Modern Concepts and Overview; Grody, W.W.; Nakamura, R.M.; Strom, C.M.; Kiechle, F.L, Eds.; Academic Press: San Diego, CA, USA, 2010; pp. 215–223.
- Jennifer H. Gunter; Marianna Kruithof-De Julio; Eugenio Zoni; Editorial: Personalized Medicine for Urological Cancers: Targeting Cancer Metabolism. Frontiers in Oncology 2022, 12, 862811, 10.3389/fonc.2022.862811.
- Ana C. Garrido-Castro; Cristina Saura; Romualdo Barroso-Sousa; Hao Guo; Eva Ciruelos; Begoña Bermejo; Joaquin Gavilá; Violeta Serra; Aleix Prat; Laia Paré; et al.Pamela CélizPatricia VillagrasaYisheng LiJennifer SavoieZhan XuCarlos L. ArteagaIan E. KropDavid B. SolitGordon B. MillsLewis C. CantleyEric P. WinerNancy U. LinJordi Rodon Phase 2 study of buparlisib (BKM120), a pan-class I PI3K inhibitor, in patients with metastatic triple-negative breast cancer. Breast Cancer Research 2020, 22, 1-13, 10.1186/s13058-020-01354-y.
- Tori Wilhoit; PharmD Jeannie M. Patrick; PharmD Megan B. May; Alpelisib: A Novel Therapy for Patients With PIK3CA-Mutated Metastatic Breast Cancer. Journal of the Advanced Practitioner in Oncology 2020, 11, 768-775, 10.6004/jadpro.2020.11.7.9.
- B. Verret; J. Cortes; T. Bachelot; F. Andre; Monica Arnedos; Efficacy of PI3K inhibitors in advanced breast cancer. Annals of Oncology 2019, 30, x12-x20, 10.1093/annonc/mdz381.
- Felix A Mensah; Jean-Pierre Blaize; Locke J Bryan; Spotlight on copanlisib and its potential in the treatment of relapsed/refractory follicular lymphoma: evidence to date. OncoTargets and Therapy 2018, ume 11, 4817-4827, 10.2147/ott.s142264.
- Hima V. Vangapandu; Nitin Jain; Varsha Gandhi; Duvelisib: a phosphoinositide-3 kinase δ/γ inhibitor for chronic lymphocytic leukemia. Expert Opinion on Investigational Drugs 2017, 26, 625-632, 10.1080/13543784.2017.1312338.
- Chan Yoon Cheah; Nathan H. Fowler; Idelalisib in the management of lymphoma. Blood 2016, 128, 331-336, 10.1182/blood-2016-02-702761.
- Jordi Rodon; Irene Brana; Lillian L Siu; Maja J De Jonge; Natasha Homji; David Mills; Emmanuelle Di Tomaso; Celine Sarr; Lucia Trandafir; Cristian Massacesi; et al.Ferry EskensJohanna C Bendell Phase I dose-escalation and -expansion study of buparlisib (BKM120), an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. Investigational New Drugs 2014, 32, 670-681, 10.1007/s10637-014-0082-9.
- Ian E Krop; Ingrid A Mayer; Vinod Ganju; Maura Dickler; Stephen Johnston; Serafin Morales; Denise A Yardley; Bohuslav Melichar; Andres Forero-Torres; Soo Chin Lee; et al.Richard de BoerKatarina PetrakovaSusanne VallentinEdith A PerezMartine PiccartMatthew EllisEric WinerSteven GendreauMika DerynckMark LacknerGallia LevyJiaheng QiuJing HePeter Schmid Pictilisib for oestrogen receptor-positive, aromatase inhibitor-resistant, advanced or metastatic breast cancer (FERGI): a randomised, double-blind, placebo-controlled, phase 2 trial. The Lancet Oncology 2016, 17, 811-821, 10.1016/s1470-2045(16)00106-6.
- Chao Liang; Xijiao Yu; Naping Xiong; Zhichang Zhang; Zhenyu Sun; Yang Dong; Pictilisib Enhances the Antitumor Effect of Doxorubicin and Prevents Tumor-Mediated Bone Destruction by Blockade of PI3K/AKT Pathway. Frontiers in Oncology 2021, 10, 615146, 10.3389/fonc.2020.615146.