The advancement of super-resolution imaging (SRI) relies on fluorescent proteins with novel photochromic properties. Using light, the reversibly switchable fluorescent proteins (RSFPs) can be converted between bright and dark states for many photocycles and their emergence has inspired the invention of advanced SRI techniques. The general photoswitching mechanism involves the chromophore cis-trans isomerization and proton transfer for negative and positive RSFPs and hydration–dehydration for decoupled RSFPs. However, a detailed understanding of these processes on ultrafast timescales (femtosecond to millisecond) is lacking, which fundamentally hinders the further development of RSFPs. For a negative RSFP such as Dronpa, the light used to induce fluorescence can often switch the protein from the on to off state; whereas for a positive RSFP like Padron, the same light can turn more proteins on. In contrast, the decoupled RSFPs can use separate light with different wavelengths for fluorescence excitation and photoswitching.
- photoswitchable fluorescent proteins
- ultrafast techniques
- cis-trans isomerization
- proton transfer
- molecular reaction dynamics
- excited state processes
- photochemistry
1. Introduction
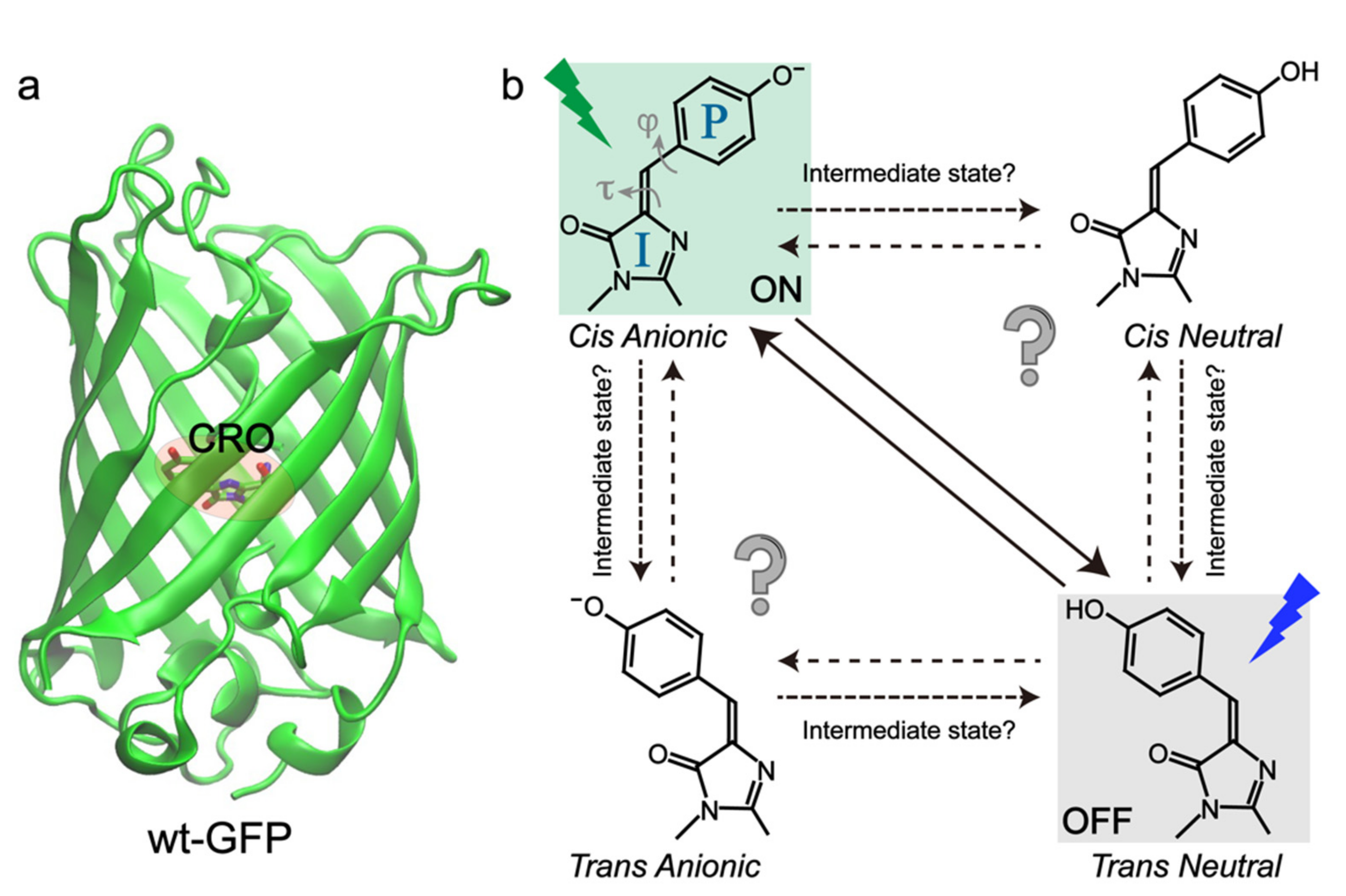
2. Negative RSFPs
2.1. Dronpa and Dronpa-2
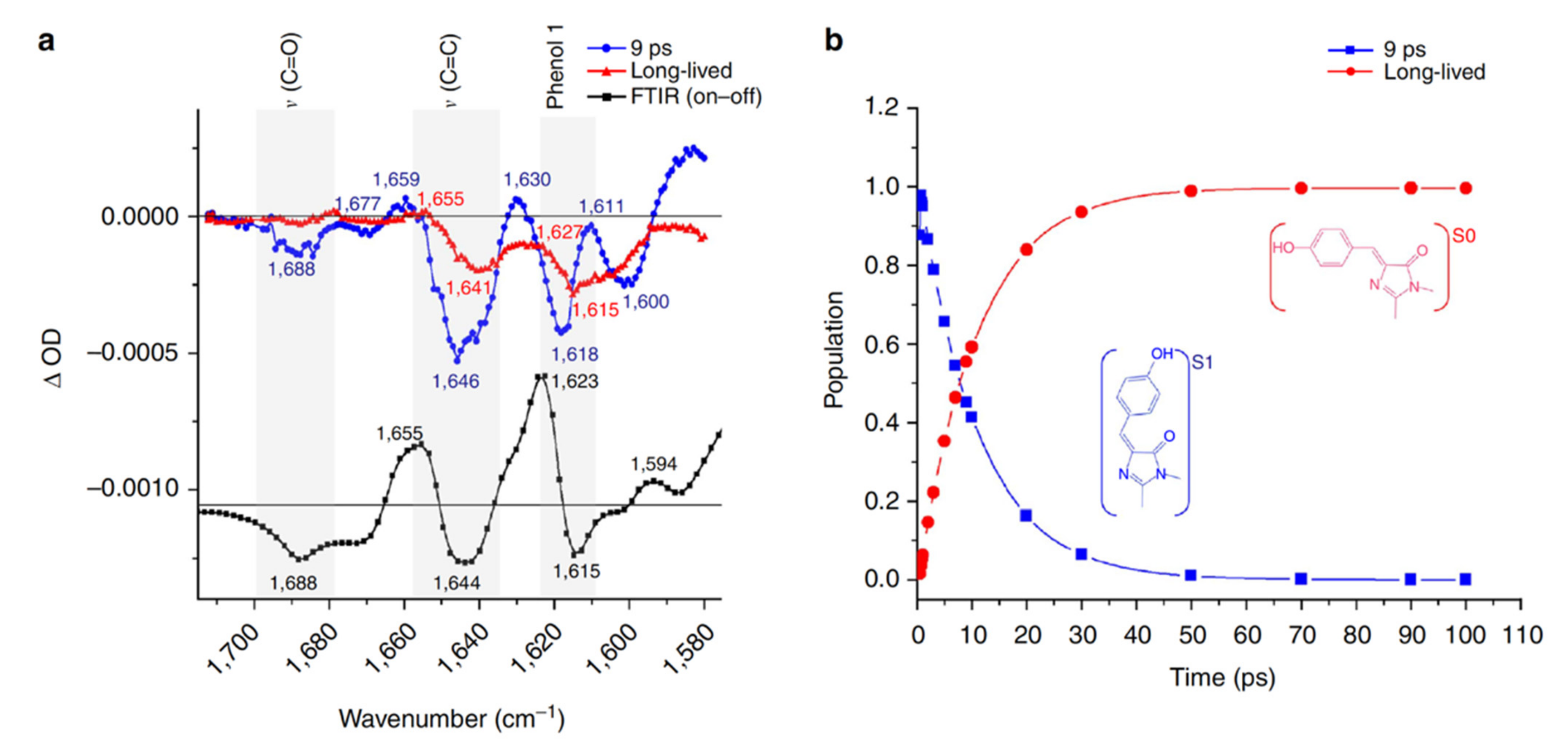
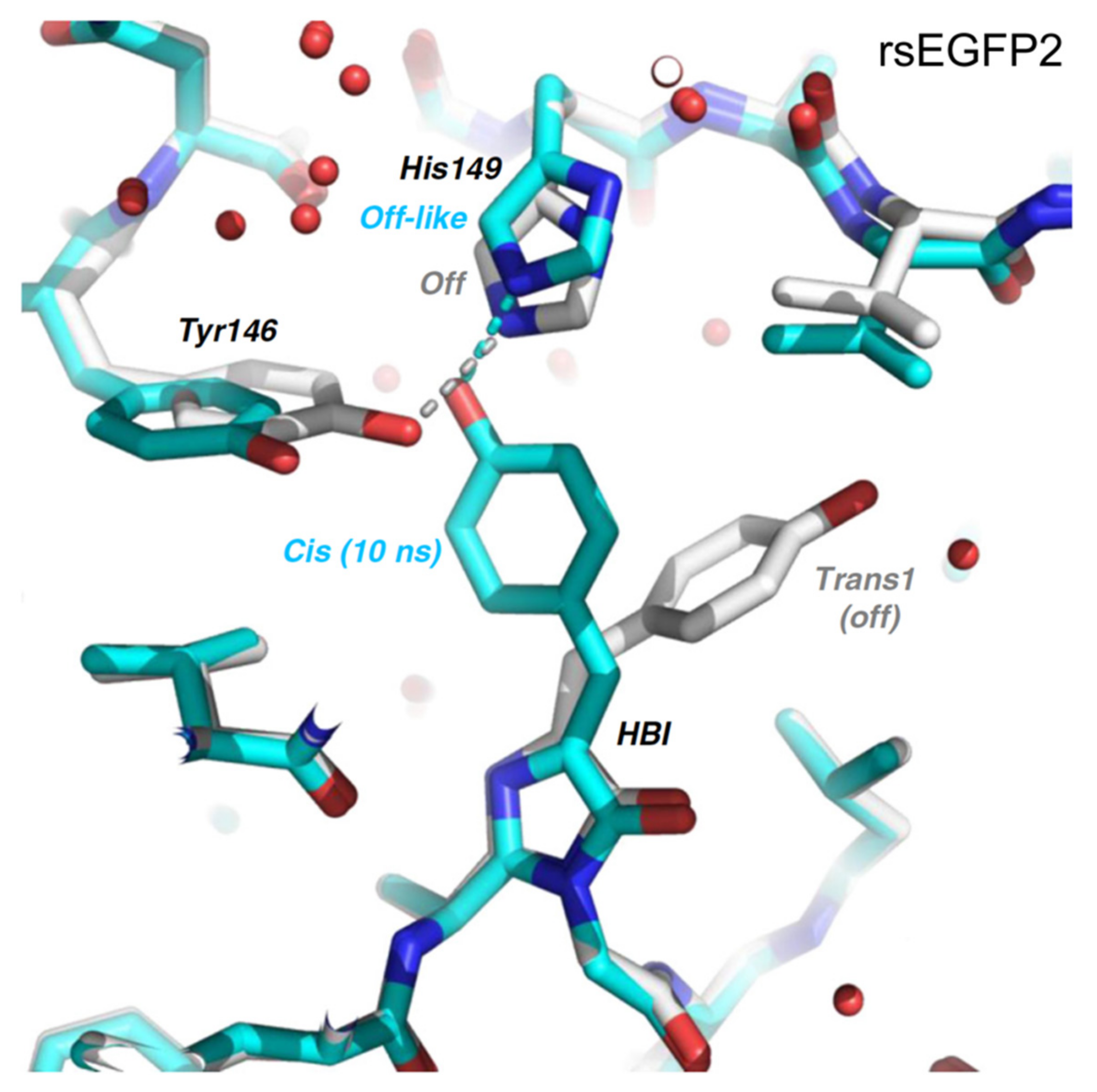
2.2. RsEGFP2
2.3. IrisFP
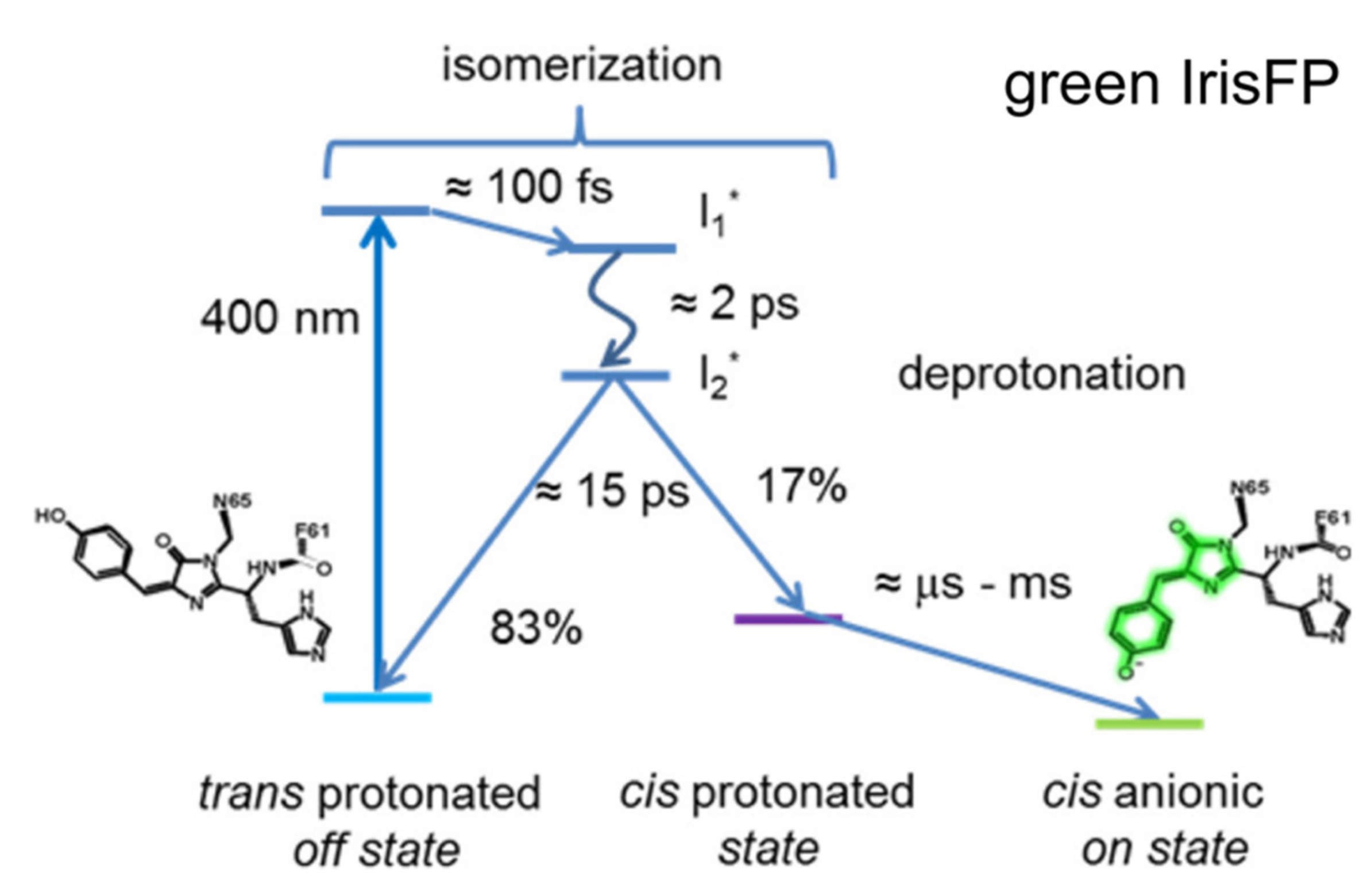
2.4. Other Negative RSFPs
Many other negative RSFPs have been engineered from teal color to the less phototoxic redder regions, such as mTFP0.7 [41][68][69], also the monomeric rsCherryRev [70], rsTagRFP (turned on by 440 nm light and off by 567 nm light, much brighter than rsCherryRev) [71], and three rsFusionRed variants (multiple irradiation wavelengths at 405/488/510 nm can be used to switch on the protein, with 590 nm light capable of switching off the protein; faster off-switching kinetics than rsTagRFP and rsCherryRev1.4) [72], by varying the chromophore composition and mutagenesis of local residues. Note that these redder proteins (than Dronpa or rsEGFP2) display some intriguing properties from multiple switching locations [72] to an unusual planar trans-chromophore that is nonfluorescent [71]. However, there have not been ultrafast studies performed on them, which represents a wonderful opportunity for researchers to implement advanced ultrafast techniques to examine these fascinating proteins. The systematic investigation and comparison of multiple negative RSFPs with contrasting properties would significantly benefit their future development in a synergistic manner. These related studies would allow the researchers to evaluate whether the retrieved ultrafast mechanisms (see above, particularly regarding the excited-state chromophore twist on the ps timescale) can be universally applied to negative RSFPs. It is expected that with a different chromophore and local protein residues, the timescales of isomerization and proton transfer would vary to different extents. However, some essential questions warrant special attention. For example, does trans-cis isomerization always occur before proton transfer? Can the researchers always see an excited-state structure that is in-between the cis and trans geometries for all RSFPs? Which local environment favors the one-bond rotation or hula twist and are these processes mutually exclusive of each other? What is the major determinant for the protonation and conformational states of the chromophore with various usual or unusual combinations of cis-/trans-/neutral/anionic/planar/nonplanar descriptors? [73] The growing pertinent comprehensive experimental findings with more advanced spectroscopic toolsets are expected to help the researchers to further understand the photoactivation mechanism from the bottom up and efficiently guide the future research directions and engineering strategies to develop next-generation negative RSFPs.References
- Chalfie, M.; Tu, Y.; Euskirchen, G.; Ward, W.W.; Prasher, D.C. Green fluorescent protein as a marker for gene expression. Science 1994, 263, 802–805.
- Tsien, R.Y. The green fluorescent protein. Annu. Rev. Biochem. 1998, 67, 509–544.
- Zimmer, M. Green fluorescent protein (GFP): Applications, structure, and related photophysical behavior. Chem. Rev. 2002, 102, 759–781.
- Jung, G. (Ed.) Fluorescent Proteins II: Application of Fluorescent Protein Technology; Springer: Berlin/Heidelberg, Germany, 2012; p. 284.
- Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.; Olenych, S.; Bonifacino, J.S.; Davidson, M.W.; Lippincott-Schwartz, J.; Hess, H.F. Imaging intracellular fluorescent proteins at nanometer resolution. Science 2006, 313, 1642–1645.
- Moerner, W.E. New directions in single-molecule imaging and analysis. Proc. Natl. Acad. Sci. USA 2007, 104, 12596–12602.
- Huang, B.; Babcock, H.; Zhuang, X. Breaking the diffraction barrier: Super-resolution imaging of cells. Cell 2010, 143, 1047–1058.
- Grotjohann, T.; Testa, I.; Leutenegger, M.; Bock, H.; Urban, N.T.; Lavoie-Cardinal, F.; Willig, K.I.; Eggeling, C.; Jakobs, S.; Hell, S.W. Diffraction-unlimited all-optical imaging and writing with a photochromic GFP. Nature 2011, 478, 204–208.
- Chozinski, T.J.; Gagnon, L.A.; Vaughan, J.C. Twinkle, twinkle little star: Photoswitchable fluorophores for super-resolution imaging. FEBS Lett. 2014, 588, 3603–3612.
- Shcherbakova, D.M.; Sengupta, P.; Lippincott-Schwartz, J.; Verkhusha, V.V. Photocontrollable fluorescent proteins for superresolution imaging. Annu. Rev. Biophys. 2014, 43, 303–329.
- Bourgeois, D.; Adam, V. Reversible photoswitching in fluorescent proteins: A mechanistic view. IUBMB Life 2012, 64, 482–491.
- Zhou, X.X.; Lin, M.Z. Photoswitchable fluorescent proteins: Ten years of colorful chemistry and exciting applications. Curr. Opin. Chem. Biol. 2013, 17, 682–690.
- Patterson, G.H.; Lippincott-Schwartz, J. A photoactivatable GFP for selective photolabeling of proteins and cells. Science 2002, 297, 1873–1877.
- Wachter, M.R. Photoconvertible fluorescent proteins and the role of dynamics in protein evolution. Int. J. Mol. Sci. 2017, 18, 1792.
- Lukyanov, K.A.; Fradkov, A.F.; Gurskaya, N.G.; Matz, M.V.; Labas, Y.A.; Savitsky, A.P.; Markelov, M.L.; Zaraisky, A.G.; Zhao, X.; Fang, Y.; et al. Natural animal coloration can be determined by a nonfluorescent green fluorescent protein homolog. J. Biol. Chem. 2000, 275, 25879–25882.
- Piatkevich, K.D.; English, B.P.; Malashkevich, V.N.; Xiao, H.; Almo, S.C.; Singer, R.H.; Verkhusha, V.V. Photoswitchable red fluorescent protein with a large Stokes shift. Chem. Biol. 2014, 21, 1402–1414.
- Zhang, X.; Zhang, M.; Li, D.; He, W.; Peng, J.; Betzig, E.; Xu, P. Highly photostable, reversibly photoswitchable fluorescent protein with high contrast ratio for live-cell superresolution microscopy. Proc. Natl. Acad. Sci. USA 2016, 113, 10364–10369.
- Pennacchietti, F.; Serebrovskaya, E.O.; Faro, A.R.; Shemyakina, I.I.; Bozhanova, N.G.; Kotlobay, A.A.; Gurskaya, N.G.; Bodén, A.; Dreier, J.; Chudakov, D.M.; et al. Fast reversibly photoswitching red fluorescent proteins for live-cell RESOLFT nanoscopy. Nat. Methods 2018, 15, 601–604.
- Wazawa, T.; Noma, R.; Uto, S.; Sugiura, K.; Washio, T.; Nagai, T. A photoswitchable fluorescent protein for hours-time-lapse and sub-second-resolved super-resolution imaging. Microscopy 2021, 70, 340–352.
- Jensen, N.A.; Jansen, I.; Kamper, M.; Jakobs, S. Reversibly switchable fluorescent proteins for RESOLFT nanoscopy. In Nanoscale Photonic Imaging; Salditt, T., Egner, A., Luke, D.R., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 241–261.
- Ando, R.; Mizuno, H.; Miyawaki, A. Regulated fast nucleocytoplasmic shuttling observed by reversible protein highlighting. Science 2004, 306, 1370–1373.
- Andresen, M.; Stiel, A.C.; Fölling, J.; Wenzel, D.; Schönle, A.; Egner, A.; Eggeling, C.; Hell, S.W.; Jakobs, S. Photoswitchable fluorescent proteins enable monochromatic multilabel imaging and dual color fluorescence nanoscopy. Nat. Biotechnol. 2008, 26, 1035–1040.
- Brakemann, T.; Stiel, A.C.; Weber, G.; Andresen, M.; Testa, I.; Grotjohann, T.; Leutenegger, M.; Plessmann, U.; Urlaub, H.; Eggeling, C.; et al. A reversibly photoswitchable GFP-like protein with fluorescence excitation decoupled from switching. Nat. Biotechnol. 2011, 29, 942–947.
- Arai, Y.; Takauchi, H.; Ogami, Y.; Fujiwara, S.; Nakano, M.; Matsuda, T.; Nagai, T. Spontaneously blinking fluorescent protein for simple single laser super-resolution live cell imaging. ACS Chem. Biol. 2018, 13, 1938–1943.
- Mats Ormö; Andrew B. Cubitt; Karen Kallio; Larry A. Gross; Roger Y. Tsien; S. James Remington; Crystal Structure of the Aequorea victoria Green Fluorescent Protein. Science 1996, 273, 1392-1395, 10.1126/science.273.5280.1392.
- Andresen, M.; Stiel, A.C.; Trowitzsch, S.; Weber, G.; Eggeling, C.; Wahl, M.C.; Hell, S.W.; Jakobs, S. Structural basis for reversible photoswitching in Dronpa. Proc. Natl. Acad. Sci. USA 2007, 104, 13005–13009.
- Wilmann, P.G.; Turcic, K.; Battad, J.M.; Wilce, M.C.J.; Devenish, R.J.; Prescott, M.; Rossjohn, J. The 1.7 Å crystal structure of Dronpa: A photoswitchable green fluorescent protein. J. Mol. Biol. 2006, 364, 213–224.
- Andre Stiel; Simon Trowitzsch; Gert Weber; Martin Andresen; Christian Eggeling; Stefan W. Hell; Stefan Jakobs; Markus Wahl; 1.8 Å bright-state structure of the reversibly switchable fluorescent protein Dronpa guides the generation of fast switching variants. Biochemical Journal 2007, 402, 35-42, 10.1042/bj20061401.
- Mizuno, H.; Mal, T.K.; Walchli, M.; Kikuchi, A.; Fukano, T.; Ando, R.; Jeyakanthan, J.; Taka, J.; Shiro, Y.; Ikura, M.; et al. Light-dependent regulation of structural flexibility in a photochromic fluorescent protein. Proc. Natl. Acad. Sci. USA 2008, 105, 9227–9232.
- Xin Li; Lung Wa Chung; Hideaki Mizuno; Atsushi Miyawaki; Keiji Morokuma; A Theoretical Study on the Nature of On- and Off-States of Reversibly Photoswitching Fluorescent Protein Dronpa: Absorption, Emission, Protonation, and Raman. The Journal of Physical Chemistry B 2009, 114, 1114-1126, 10.1021/jp909947c.
- Daryna Smyrnova; Kirill Zinovjev; Iñaki Tuñón; Arnout Ceulemans; Thermal Isomerization Mechanism in Dronpa and Its Mutants. The Journal of Physical Chemistry B 2016, 120, 12820-12825, 10.1021/acs.jpcb.6b10859.
- Yen-Cheng Chen; Amy E. Jablonski; Irina Issaeva; Daisy Bourassa; Jung-Cheng Hsiang; Christoph J. Fahrni; Robert M. Dickson; Optically Modulated Photoswitchable Fluorescent Proteins Yield Improved Biological Imaging Sensitivity. Journal of the American Chemical Society 2015, 137, 12764-12767, 10.1021/jacs.5b07871.
- Ryoko Ando; Cristina Flors; Hideaki Mizuno; Johan Hofkens; Atsushi Miyawaki; Highlighted Generation of Fluorescence Signals Using Simultaneous Two-Color Irradiation on Dronpa Mutants. Biophysical Journal 2007, 92, L97-L99, 10.1529/biophysj.107.105882.
- Chong Fang; Longteng Tang; Cheng Chen; Unveiling coupled electronic and vibrational motions of chromophores in condensed phases. The Journal of Chemical Physics 2019, 151, 200901, 10.1063/1.5128388.
- Satoshi Habuchi; Ryoko Ando; Peter Dedecker; Wendy Verheijen; Hideaki Mizuno; Atsushi Miyawaki; Johan Hofkens; Reversible single-molecule photoswitching in the GFP-like fluorescent protein Dronpa. Proceedings of the National Academy of Sciences 2005, 102, 9511-9516, 10.1073/pnas.0500489102.
- Eduard Fron; Cristina Flors; Gerd Schweitzer; Satoshi Habuchi; Hideaki Mizuno; Ryoko Ando; Frans C. De Schryver; Atsushi Miyawaki; Johan Hofkens; Ultrafast Excited-State Dynamics of the Photoswitchable Protein Dronpa. Journal of the American Chemical Society 2007, 129, 4870-4871, 10.1021/ja069365v.
- Satoshi Habuchi; Ryoko Ando; Peter Dedecker; Wendy Verheijen; Hideaki Mizuno; Atsushi Miyawaki; Johan Hofkens; Reversible single-molecule photoswitching in the GFP-like fluorescent protein Dronpa. Proceedings of the National Academy of Sciences 2005, 102, 9511-9516, 10.1073/pnas.0500489102.
- Mark M. Warren; Marius Kaucikas; Ann Fitzpatrick; Paul Champion; J. Timothy Sage; Jasper J. Van Thor; Ground-state proton transfer in the photoswitching reactions of the fluorescent protein Dronpa. Nature Communications 2013, 4, 1461, 10.1038/ncomms2460.
- Andras Lukacs; Allison Haigney; Richard Brust; Kiri Addison; Michael Towrie; Gregory M. Greetham; Garth A. Jones; Atsushi Miyawaki; Peter J. Tonge; Stephen R. Meech; et al. Protein Photochromism Observed by Ultrafast Vibrational Spectroscopy. The Journal of Physical Chemistry B 2013, 117, 11954-11959, 10.1021/jp406142g.
- Chong Fang; Renee Frontiera; Rosalie Tran; Richard A. Mathies; Mapping GFP structure evolution during proton transfer with femtosecond Raman spectroscopy. Nature 2009, 462, 200-204, 10.1038/nature08527.
- J. Nathan Henderson; Hui-Wang Ai; Robert E. Campbell; S. James Remington; Structural basis for reversible photobleaching of a green fluorescent protein homologue. Proceedings of the National Academy of Sciences 2007, 104, 6672-6677, 10.1073/pnas.0700059104.
- Fangyuan Han; Weimin Liu; Chong Fang; Excited-state proton transfer of photoexcited pyranine in water observed by femtosecond stimulated Raman spectroscopy. Chemical Physics 2013, 422, 204-219, 10.1016/j.chemphys.2013.03.009.
- Ando, R.; Flors, C.; Mizuno, H.; Hofkens, J.; Miyawaki, A. Highlighted generation of fluorescence signals using simultaneous two-color irradiation on Dronpa mutants. Biophys. J. 2007, 92, L97–L99.
- Yadav, D.; Lacombat, F.; Dozova, N.; Rappaport, F.; Plaza, P.; Espagne, A. Real-time monitoring of chromophore isomerization and deprotonation during the photoactivation of the fluorescent protein Dronpa. J. Phys. Chem. B 2015, 119, 2404–2414.
- Kaucikas, M.; Tros, M.; van Thor, J.J. Photoisomerization and proton transfer in the forward and reverse photoswitching of the fast-switching M159T mutant of the Dronpa fluorescent protein. J. Phys. Chem. B 2015, 119, 2350–2362.
- David W. McCamant; Philipp Kukura; Sangwoon Yoon; Richard A. Mathies; Femtosecond broadband stimulated Raman spectroscopy: Apparatus and methods. Review of Scientific Instruments 2004, 75, 4971-4980, 10.1063/1.1807566.
- Fang, C.; Tang, L. Mapping structural dynamics of proteins with femtosecond stimulated Raman spectroscopy. Annu. Rev. Phys. Chem. 2020, 71, 239–265.
- Nicolas Coquelle; Michel Sliwa; Joyce Woodhouse; Giorgio Schiro; Virgile Adam; Andrew Aquila; Thomas R. M. Barends; Sébastien Boutet; Martin Byrdin; Sergio Carbajo; et al.Eugenio de la MoraR. Bruce DoakMikolaj FeliksFranck FieschiLutz FoucarVirginia GuillonMario HilpertMark S. HunterStefan JakobsJason E. KoglinGabriela KovacsovaThomas LaneBernard LévyMengning LiangKarol NassJacqueline RidardJoseph S. RobinsonChristopher M. RoomeCyril RuckebuschMatthew SeabergMichel ThepautMarco CammarataIsabelle DemachyMartin J FieldRobert L. ShoemanDominique BourgeoisJacques-Philippe ColletierIlme SchlichtingMartin Weik Chromophore twisting in the excited state of a photoswitchable fluorescent protein captured by time-resolved serial femtosecond crystallography. Nature Chemistry 2017, 10, 31-37, 10.1038/nchem.2853.
- Smyrnova, D.; Zinovjev, K.; Tuñón, I.; Ceulemans, A. Thermal isomerization mechanism in Dronpa and its mutants. J. Phys. Chem. B 2016, 120, 12820–12825.
- Samer Gozem; Hoi Ling Luk; Igor Schapiro; Massimo Olivucci; Theory and Simulation of the Ultrafast Double-Bond Isomerization of Biological Chromophores. Chemical Reviews 2017, 117, 13502-13565, 10.1021/acs.chemrev.7b00177.
- Shimomura, O.; Johnson, F.H.; Saiga, Y. Extraction, purification and properties of Aequorin, a bioluminescent protein from the luminous Hydromedusan, Aequorea. J. Cell. Comp. Physiol. 1962, 59, 223–239.
- Brejc, K.; Sixma, T.K.; Kitts, P.A.; Kain, S.R.; Tsien, R.Y.; Ormö, M.; Remington, S.J. Structural basis for dual excitation and photoisomerization of the Aequorea victoria green fluorescent protein. Proc. Natl. Acad. Sci. USA 1997, 94, 2306–2311.
- Cormack, B.P.; Valdivia, R.H.; Falkow, S. FACS-optimized mutants of the green fluorescent protein (GFP). Gene 1996, 173, 33–38.
- Grotjohann, T.; Testa, I.; Reuss, M.; Brakemann, T.; Eggeling, C.; Hell, S.W.; Jakobs, S. RsEGFP2 enables fast RESOLFT nanoscopy of living cells. eLife 2012, 1, e00248.
- Mariam El Khatib; Alexandre Martins; Dominique Bourgeois; Jacques-Philippe Colletier; Virgile Adam; Rational design of ultrastable and reversibly photoswitchable fluorescent proteins for super-resolution imaging of the bacterial periplasm. Scientific Reports 2016, 6, srep18459, 10.1038/srep18459.
- Coquelle, N.; Sliwa, M.; Woodhouse, J.; Schirò, G.; Adam, V.; Aquila, A.; Barends, T.R.M.; Boutet, S.; Byrdin, M.; Carbajo, S.; et al. Chromophore twisting in the excited state of a photoswitchable fluorescent protein captured by time-resolved serial femtosecond crystallography. Nat. Chem. 2018, 10, 31–37.
- Uriarte, L.M.; Vitale, R.; Niziński, S.; Hadjidemetriou, K.; Zala, N.; Lukacs, A.; Greetham, G.M.; Sazanovich, I.V.; Weik, M.; Ruckebusch, C.; et al. Structural information about the trans-to-cis isomerization mechanism of the photoswitchable fluorescent protein rsEGFP2 revealed by multiscale infrared transient absorption. J. Phys. Chem. Lett. 2022, 13, 1194–1202.
- Jeffrey Chang; Matthew G. Romei; Steven G. Boxer; Structural Evidence of Photoisomerization Pathways in Fluorescent Proteins. Journal of the American Chemical Society 2019, 141, 15504-15508, 10.1021/jacs.9b08356.
- Virgile Adam; Kyprianos Hadjidemetriou; Nickels Jensen; Robert L. Shoeman; Joyce Woodhouse; Andrew Aquila; Anne-Sophie Banneville; Thomas R. M. Barends; Victor Bezchastnov; Sébastien Boutet; et al.Martin ByrdinMarco CammarataSergio CarbajoNina Eleni ChristouNicolas CoquelleEugenio De la MoraMariam El KhatibTadeo Moreno ChicanoR. Bruce DoakFranck FieschiLutz FoucarOleksandr GlushonkovAlexander GorelMarie Luise GrünbeinMario HilpertMark HunterMarco KloosJason E. KoglinThomas J. LaneMengning LiangAngela MantovanelliKarol NassGabriela Nass KovacsShigeki OwadaChristopher M. RoomeGiorgio SchiròMatthew SeabergMiriam StrickerMichel ThépautKensuke TonoKiyoshi UedaLucas M. UriarteDaehyun YouNinon ZalaTatiana DomratchevaStefan JakobsMichel SliwaIlme SchlichtingJacques-Philippe ColletierDominique BourgeoisMartin Weik Rational control of structural off-state heterogeneity in a photoswitchable fluorescent protein provides switching contrast enhancement. bioRxiv 2021, New_Results, DOI: https://doi.org/10.1101/2021.11.05.462999, 10.1101/2021.11.05.462999.
- Woodhouse, J.; Nass Kovacs, G.; Coquelle, N.; Uriarte, L.M.; Adam, V.; Barends, T.R.M.; Byrdin, M.; de la Mora, E.; Bruce Doak, R.; Feliks, M.; et al. Photoswitching mechanism of a fluorescent protein revealed by time-resolved crystallography and transient absorption spectroscopy. Nat. Commun. 2020, 11, 741.
- Laptenok, S.P.; Gil, A.A.; Hall, C.R.; Lukacs, A.; Iuliano, J.N.; Jones, G.A.; Greetham, G.M.; Donaldson, P.; Miyawaki, A.; Tonge, P.J.; et al. Infrared spectroscopy reveals multi-step multi-timescale photoactivation in the photoconvertible protein archetype Dronpa. Nat. Chem. 2018, 10, 845–852.
- Jörg Wiedenmann; Sergey Ivanchenko; Franz Oswald; Florian Schmitt; Carlheinz Röcker; Anya Salih; Klaus-Dieter Spindler; G. Ulrich Nienhaus; EosFP, a fluorescent marker protein with UV-inducible green-to-red fluorescence conversion. Proceedings of the National Academy of Sciences 2004, 101, 15905-15910, 10.1073/pnas.0403668101.
- Adam, V.; Lelimousin, M.; Boehme, S.; Desfonds, G.; Nienhaus, K.; Field, M.J.; Wiedenmann, J.; McSweeney, S.; Nienhaus, G.U.; Bourgeois, D. Structural characterization of IrisFP, an optical highlighter undergoing multiple photo-induced transformations. Proc. Natl. Acad. Sci. USA 2008, 105, 18343–18348.
- Jacques-Philippe Colletier; Michel Sliwa; François-Xavier Gallat; Michihiro Sugahara; Virginia Guillon; Giorgio Schirò; Nicolas Coquelle; Joyce Woodhouse; Laure Roux; Guillaume Gotthard; et al.Antoine RoyantLucas Martinez UriarteCyril RuckebuschYasumasa JotiMartin ByrdinEiichi MizohataEriko NangoTomoyuki TanakaKensuke TonoMakina YabashiVirgile AdamMarco CammarataIlme SchlichtingDominique BourgeoisMartin Weik Serial Femtosecond Crystallography and Ultrafast Absorption Spectroscopy of the Photoswitchable Fluorescent Protein IrisFP. The Journal of Physical Chemistry Letters 2016, 7, 882-887, 10.1021/acs.jpclett.5b02789.
- Colletier, J.-P.; Sliwa, M.; Gallat, F.-X.; Sugahara, M.; Guillon, V.; Schirò, G.; Coquelle, N.; Woodhouse, J.; Roux, L.; Gotthard, G.; et al. Serial femtosecond crystallography and ultrafast absorption spectroscopy of the photoswitchable fluorescent protein IrisFP. J. Phys. Chem. Lett. 2016, 7, 882–887.
- Krueger, T.D.; Tang, L.; Zhu, L.; Breen, I.L.; Wachter, R.M.; Fang, C. Dual illumination enhances transformation of an engineered green-to-red photoconvertible fluorescent protein. Angew. Chem. Int. Ed. 2020, 59, 1644–1652.
- Kim, H.; Zou, T.; Modi, C.; Dörner, K.; Grunkemeyer, T.J.; Chen, L.; Fromme, R.; Matz, M.V.; Ozkan, S.B.; Wachter, R.M. A hinge migration mechanism unlocks the evolution of green-to-red photoconversion in GFP-like proteins. Structure 2015, 23, 34–43.
- Hui-Wang Ai; J. Nathan Henderson; S. James Remington; Robert E. Campbell; Directed evolution of a monomeric, bright and photostable version of Clavularia cyan fluorescent protein: structural characterization and applications in fluorescence imaging. Biochemical Journal 2006, 400, 531-540, 10.1042/bj20060874.
- Hui-Wang Ai; Robert E. Campbell; Teal fluorescent proteins: characterization of a reversibly photoswitchable variant. Biomedical Optics (BiOS) 2008 2008, 6868, 68680D-68680D-7, 10.1117/12.761423.
- Andre C. Stiel; Martin Andresen; Hannes Bock; Michael Hilbert; Jessica Schilde; Andreas Schönle; Christian Eggeling; Alexander Egner; Stefan W. Hell; Stefan Jakobs; et al. Generation of Monomeric Reversibly Switchable Red Fluorescent Proteins for Far-Field Fluorescence Nanoscopy. Biophysical Journal 2008, 95, 2989-2997, 10.1529/biophysj.108.130146.
- Sergei Pletnev; Fedor V. Subach; Zbigniew Dauter; Alexander Wlodawer; Vladislav V. Verkhusha; A Structural Basis for Reversible Photoswitching of Absorbance Spectra in Red Fluorescent Protein rsTagRFP. Journal of Molecular Biology 2012, 417, 144-151, 10.1016/j.jmb.2012.01.044.
- Francesca Pennacchietti; Ekaterina O. Serebrovskaya; Aline R. Faro; Irina I. Shemyakina; Nina G. Bozhanova; Alexey A. Kotlobay; Nadya G. Gurskaya; Andreas Bodén; Jes Dreier; Dmitry M. Chudakov; et al.Konstantin LukyanovVladislav VerkhushaAlexander S. MishinIlaria Testa Fast reversibly photoswitching red fluorescent proteins for live-cell RESOLFT nanoscopy. Nature Methods 2018, 15, 601-604, 10.1038/s41592-018-0052-9.
- Longteng Tang; Chong Fang; Fluorescence Modulation by Ultrafast Chromophore Twisting Events: Developing a Powerful Toolset for Fluorescent-Protein-Based Imaging. The Journal of Physical Chemistry B 2021, 125, 13610-13623, 10.1021/acs.jpcb.1c08570.
- Longteng Tang; Chong Fang; Fluorescence Modulation by Ultrafast Chromophore Twisting Events: Developing a Powerful Toolset for Fluorescent-Protein-Based Imaging. The Journal of Physical Chemistry B 2021, 125, 13610-13623, 10.1021/acs.jpcb.1c08570.
- Longteng Tang; Chong Fang; Fluorescence Modulation by Ultrafast Chromophore Twisting Events: Developing a Powerful Toolset for Fluorescent-Protein-Based Imaging. The Journal of Physical Chemistry B 2021, 125, 13610-13623, 10.1021/acs.jpcb.1c08570.
- Longteng Tang; Chong Fang; Fluorescence Modulation by Ultrafast Chromophore Twisting Events: Developing a Powerful Toolset for Fluorescent-Protein-Based Imaging. The Journal of Physical Chemistry B 2021, 125, 13610-13623, 10.1021/acs.jpcb.1c08570.
- Longteng Tang; Chong Fang; Fluorescence Modulation by Ultrafast Chromophore Twisting Events: Developing a Powerful Toolset for Fluorescent-Protein-Based Imaging. The Journal of Physical Chemistry B 2021, 125, 13610-13623, 10.1021/acs.jpcb.1c08570.
- Longteng Tang; Chong Fang; Fluorescence Modulation by Ultrafast Chromophore Twisting Events: Developing a Powerful Toolset for Fluorescent-Protein-Based Imaging. The Journal of Physical Chemistry B 2021, 125, 13610-13623, 10.1021/acs.jpcb.1c08570.
- Longteng Tang; Chong Fang; Fluorescence Modulation by Ultrafast Chromophore Twisting Events: Developing a Powerful Toolset for Fluorescent-Protein-Based Imaging. The Journal of Physical Chemistry B 2021, 125, 13610-13623, 10.1021/acs.jpcb.1c08570.
- Francesca Pennacchietti; Ekaterina O. Serebrovskaya; Aline R. Faro; Irina I. Shemyakina; Nina G. Bozhanova; Alexey A. Kotlobay; Nadya G. Gurskaya; Andreas Bodén; Jes Dreier; Dmitry M. Chudakov; et al.Konstantin LukyanovVladislav VerkhushaAlexander S. MishinIlaria Testa Fast reversibly photoswitching red fluorescent proteins for live-cell RESOLFT nanoscopy. Nature Methods 2018, 15, 601-604, 10.1038/s41592-018-0052-9.
- Longteng Tang; Chong Fang; Fluorescence Modulation by Ultrafast Chromophore Twisting Events: Developing a Powerful Toolset for Fluorescent-Protein-Based Imaging. The Journal of Physical Chemistry B 2021, 125, 13610-13623, 10.1021/acs.jpcb.1c08570.
- Ryoko Ando; Cristina Flors; Hideaki Mizuno; Johan Hofkens; Atsushi Miyawaki; Highlighted Generation of Fluorescence Signals Using Simultaneous Two-Color Irradiation on Dronpa Mutants. Biophysical Journal 2007, 92, L97-L99, 10.1529/biophysj.107.105882.
- Satoshi Habuchi; Ryoko Ando; Peter Dedecker; Wendy Verheijen; Hideaki Mizuno; Atsushi Miyawaki; Johan Hofkens; Reversible single-molecule photoswitching in the GFP-like fluorescent protein Dronpa. Proceedings of the National Academy of Sciences 2005, 102, 9511-9516, 10.1073/pnas.0500489102.
- Eduard Fron; Cristina Flors; Gerd Schweitzer; Satoshi Habuchi; Hideaki Mizuno; Ryoko Ando; Frans C. De Schryver; Atsushi Miyawaki; Johan Hofkens; Ultrafast Excited-State Dynamics of the Photoswitchable Protein Dronpa. Journal of the American Chemical Society 2007, 129, 4870-4871, 10.1021/ja069365v.
- Mats Ormö; Andrew B. Cubitt; Karen Kallio; Larry A. Gross; Roger Y. Tsien; S. James Remington; Crystal Structure of the Aequorea victoria Green Fluorescent Protein. Science 1996, 273, 1392-1395, 10.1126/science.273.5280.1392.
- Pascal Wilmann; Kristina Turcic; Jion M. Battad; Matthew C.J. Wilce; Rodney J. Devenish; Mark Prescott; Jamie Rossjohn; The 1.7 Å Crystal Structure of Dronpa: A Photoswitchable Green Fluorescent Protein. Journal of Molecular Biology 2006, 364, 213-224, 10.1016/j.jmb.2006.08.089.
- Andre Stiel; Simon Trowitzsch; Gert Weber; Martin Andresen; Christian Eggeling; Stefan W. Hell; Stefan Jakobs; Markus Wahl; 1.8 Å bright-state structure of the reversibly switchable fluorescent protein Dronpa guides the generation of fast switching variants. Biochemical Journal 2007, 402, 35-42, 10.1042/bj20061401.
- Daryna Smyrnova; Kirill Zinovjev; Iñaki Tuñón; Arnout Ceulemans; Thermal Isomerization Mechanism in Dronpa and Its Mutants. The Journal of Physical Chemistry B 2016, 120, 12820-12825, 10.1021/acs.jpcb.6b10859.
- Andre Stiel; Simon Trowitzsch; Gert Weber; Martin Andresen; Christian Eggeling; Stefan W. Hell; Stefan Jakobs; Markus Wahl; 1.8 Å bright-state structure of the reversibly switchable fluorescent protein Dronpa guides the generation of fast switching variants. Biochemical Journal 2007, 402, 35-42, 10.1042/bj20061401.
- Yen-Cheng Chen; Amy E. Jablonski; Irina Issaeva; Daisy Bourassa; Jung-Cheng Hsiang; Christoph J. Fahrni; Robert M. Dickson; Optically Modulated Photoswitchable Fluorescent Proteins Yield Improved Biological Imaging Sensitivity. Journal of the American Chemical Society 2015, 137, 12764-12767, 10.1021/jacs.5b07871.
- Andre Stiel; Simon Trowitzsch; Gert Weber; Martin Andresen; Christian Eggeling; Stefan W. Hell; Stefan Jakobs; Markus Wahl; 1.8 Å bright-state structure of the reversibly switchable fluorescent protein Dronpa guides the generation of fast switching variants. Biochemical Journal 2007, 402, 35-42, 10.1042/bj20061401.
- Mark M. Warren; Marius Kaucikas; Ann Fitzpatrick; Paul Champion; J. Timothy Sage; Jasper J. Van Thor; Ground-state proton transfer in the photoswitching reactions of the fluorescent protein Dronpa. Nature Communications 2013, 4, 1461, 10.1038/ncomms2460.
- Andras Lukacs; Allison Haigney; Richard Brust; Kiri Addison; Michael Towrie; Gregory M. Greetham; Garth A. Jones; Atsushi Miyawaki; Peter J. Tonge; Stephen R. Meech; et al. Protein Photochromism Observed by Ultrafast Vibrational Spectroscopy. The Journal of Physical Chemistry B 2013, 117, 11954-11959, 10.1021/jp406142g.
- Chong Fang; Renee Frontiera; Rosalie Tran; Richard A. Mathies; Mapping GFP structure evolution during proton transfer with femtosecond Raman spectroscopy. Nature 2009, 462, 200-204, 10.1038/nature08527.
- J. Nathan Henderson; Hui-Wang Ai; Robert E. Campbell; S. James Remington; Structural basis for reversible photobleaching of a green fluorescent protein homologue. Proceedings of the National Academy of Sciences 2007, 104, 6672-6677, 10.1073/pnas.0700059104.
- Fangyuan Han; Weimin Liu; Chong Fang; Excited-state proton transfer of photoexcited pyranine in water observed by femtosecond stimulated Raman spectroscopy. Chemical Physics 2013, 422, 204-219, 10.1016/j.chemphys.2013.03.009.
- Nicolas Coquelle; Michel Sliwa; Joyce Woodhouse; Giorgio Schiro; Virgile Adam; Andrew Aquila; Thomas R. M. Barends; Sébastien Boutet; Martin Byrdin; Sergio Carbajo; et al.Eugenio de la MoraR. Bruce DoakMikolaj FeliksFranck FieschiLutz FoucarVirginia GuillonMario HilpertMark S. HunterStefan JakobsJason E. KoglinGabriela KovacsovaThomas LaneBernard LévyMengning LiangKarol NassJacqueline RidardJoseph S. RobinsonChristopher M. RoomeCyril RuckebuschMatthew SeabergMichel ThepautMarco CammarataIsabelle DemachyMartin J FieldRobert L. ShoemanDominique BourgeoisJacques-Philippe ColletierIlme SchlichtingMartin Weik Chromophore twisting in the excited state of a photoswitchable fluorescent protein captured by time-resolved serial femtosecond crystallography. Nature Chemistry 2017, 10, 31-37, 10.1038/nchem.2853.
- Samer Gozem; Hoi Ling Luk; Igor Schapiro; Massimo Olivucci; Theory and Simulation of the Ultrafast Double-Bond Isomerization of Biological Chromophores. Chemical Reviews 2017, 117, 13502-13565, 10.1021/acs.chemrev.7b00177.
- Hui-Wang Ai; J. Nathan Henderson; S. James Remington; Robert E. Campbell; Directed evolution of a monomeric, bright and photostable version of Clavularia cyan fluorescent protein: structural characterization and applications in fluorescence imaging. Biochemical Journal 2006, 400, 531-540, 10.1042/bj20060874.
- Hui-Wang Ai; Robert E. Campbell; Teal fluorescent proteins: characterization of a reversibly photoswitchable variant. Biomedical Optics (BiOS) 2008 2008, 6868, 68680D-68680D-7, 10.1117/12.761423.