Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Zvonimir Bosnic and Version 2 by Sirius Huang.
Inflammation constitutes an essential response of tissues to infection or injury. The cytokine IL-37 is a member of the IL-1 family of cytokines, otherwise known for its pivotal role in promoting inflammation. This cytokine (a former member 7 of the IL-1 family) was characterized by computational cloning, which revealed its role as a negative regulator of the cytokine IL-18, which in addition to IL-1β is the key proinflammatory cytokine of the IL-1 cytokine family. There is growing interest in the therapeutic potentials of IL-37.
- chronic inflammation
- cardio-metabolic disorders
- cytokine IL-37
- thyroid hormones
1. The Role of the Cytokine IL-37 in Regulating Acute Inflammation
1.1. Innate Immunity and Acute Inflammation
As a result of the fact that IL-1β and IL-1α, the first members of the IL-1 cytokine family to be discovered, were found to have proinflammatory properties, the whole IL-1 cytokine family was thought to have a role in amplifying innate immune and inflammatory responses. Technological advances in the last few decades have enabled the discovery of new cytokines of this family, with both pro- and anti-inflammatory properties [1][75]. It has been further determined that the IL-1 receptor (IL-1R) family members not only have ligand-binding chains, but also contain molecules that act as negative regulators of cellular signaling [2][76]. Today, after all molecules of the IL-1 family have been identified and classified according to the unique nomenclature, we can better understand the principles of functioning of this signaling system. Owing to its strong proinflammatory properties, this system also possesses mechanisms of self-control and fine-tuning of proinflammatory effects, the aim of which is to preserve tissues at the site of infection (inflammation) from unnecessary collateral damage and autoimmune reactions [3][77]. Molecules of the IL-1 cytokine family, which function to maintain control over unrestrained inflammation, include soluble molecules, such as IL-1 and IL-36 receptor antagonists (IL-1Ra and IL-36Ra), suppressor cytokines IL-37 and IL-38, and receptor-inhibiting molecules, such as IL-18 binding protein (IL-18BP) [3][4][74,77]. In addition, there are many checkpoints along the IL-1-dependent intracellular signaling pathways where variations in substrate concentrations or changes in component switch or cleavage can serve as limitation factors and can inhibit propagation of the proinflammatory signals [3][77].
In principle, when cytokines IL-1β or IL-1α bind to their IL-1R1 expressed on the surface of innate immune cells such as macrophages and antigen-presenting DCs, proinflammatory signals are initiated within these cells [5][72]. IL-1R1 is a ligand-binding receptor molecule. After ligation to its agonist, this molecule undergoes structural changes, which allows the coreceptor molecule IL-1R3 to approach and form the functional tripartite receptor complex. Intracellular signaling proceeds via intracellular domains of these two receptor components, which are structurally similar to intracellular domains of the Toll-like innate immune receptor and are therefore termed Toll IL-1R (TIR) domains. When TIR domains approach each other, conditions are created for binding to the adaptor molecule MyD88, whereby this molecule becomes phosphorylated and capable of initiating the canonical activation signal to intracellular kinases (IRAKs) [1][5][72,75]. This is an essential proinflammatory pathway mediated by IL-1β as the prototypic proinflammatory cytokine and involves the proteins TRAF6 and TAK1 and IKK kinases, leading to activation of the transcription factor nuclear factor kappa B (NF-κB) [5][72]. This transcription factor is known for its pivotal role in regulating a large array of genes involved in inflammatory and immune responses (Figure 1) [6][78]. Among the most important of these genes are those that code proinflammatory cytokines, for example, tumor necrosis factor alpha (TNF-α), IL-1α, IL-β, IL-6, IL-12, IL-15, and IL-18; those that code chemokines (inflammatory cell chemoattractant factors), such as IL-8, monocyte chemoattractant protein-1 (MCP-1), and granulocyte- and granulocyte-macrophage colony stimulating factor (G- and GM-CSF); those that code adhesion molecules, such as intercellular adhesion molecule 1 (ICAM-1); those that regulate the survival, activation, and differentiation of innate immune cells and T lymphocytes; and those that participate in inflammasome regulation (the intracellular multi-protein activators of caspases—enzymes involved in lytic programmed cell death or pyroptosis) (Figure 1) [5][6][7][8][72,78,79,80]. Thus, the resulting cellular activities include gene transcription, translation processes, autophagy (degradation of damaged or redundant cellular components through lysosome-dependent regulated mechanisms), pyroptosis (a caspase-dependent inflammation-mediated lytic cell death), necroptosis (a caspase-independent inflammation-mediated lytic cell death), immune-mediated metabolic changes, and oxidative phosphorylation of enzymes participating in signaling pathways (Figure 1) [3][77].
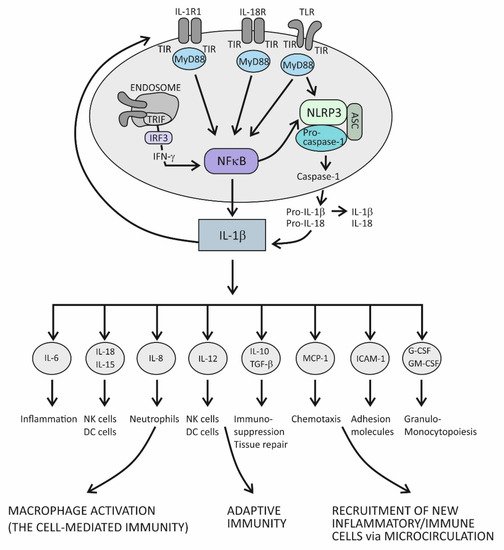
Figure 1.
The role of the IL-1 cytokine family in promoting inflammation.
The role of the IL-1 family of molecules, in fact, is to amplify or fine-tune the innate immune responses on antigens recognized by innate immune receptors. These receptors, such as Toll-like receptors (TLRs), are displayed on the surface of innate immune cells, including mainly macrophages, DCs, and neutrophils, or are positioned intracellularly, in the proximity of inflammasomes (the intracellular sensing system), such as NOD-like receptors (also termed nucleotide-binding domain and leucine-rich repeat pyrin-containing protein receptors) (NLRs) (Figure 1) [1][2][3][4][7][8][9][10][73,74,75,76,77,79,80,81]. By using the innate immune receptors, innate immune cells search the microenvironment for the presence of PAMPs or DAMPs. As molecules of the IL-1 and TLR families share similar functions in providing innate immune responses, these two signaling systems share the same group of cytosolic domains—TIR domains [3][5][72,77]. This way, by using the prepared intracellular signaling components, the proinflammatory cytokines of the IL-1 family can amplify inflammatory signals initially generated by TLRs (Figure 1) [3][77].
There is another important inflammation amplification pathway with prolonged effects. It involves new transcriptions of the precursor molecules of the proinflammatory cytokines IL-1β and IL-18 and inflammasome receptor component NLRP3 (Figure 1) [3][5][72,77]. Several types of inflammasomes have been identified in humans, but the one containing the NLRP3 receptor has been examined best, as it plays a pivotal role in human physiology [7][8][79,80]. In addition to the receptor component, inflammasome complexes also comprise an adaptor protein ASC and an executioner component—an enzyme from the caspase family, notably, caspase-1. The role of the NLRP3 complex is to process the precursor proinflammatory cytokines IL-1β and IL-18 into mature, more active forms. The execution program involves activating the proteolytic enzyme caspase-1 and is tightly controlled by the canonical reaction, which starts with the assembly of particular NLRP3 inflammasome components and is followed by the recruitment and activation of pro-caspase 1, finally leading to proteolytical modification of the IL-1β and IL-18 precursor forms [3][8][10][77,80,81]. Once activated, caspase-1 also proteolytically modifies the membrane protein gasderin-D, which then dimerizes, forming large pores in the cell membrane through which the mature forms of the cytokines IL-1β and IL-18 are expelled from the cell into the extracellular space (Figure 1) [11][82]. Formation of the pores may eventually cause cell membrane rupture and cell lysis, leading to the release of large quantities of cytokines and other inflammatory active molecules from the cell. Additional ways by which the proinflammatory cytokines amplify inflammation involve processes such as the dispersion of activation signals to other, nonimmune cell types, including endothelial, epithelial, and mesenchymal cells, and the recruitment of new inflammatory and immune cells to the site of inflammation via dysfunctional endothelial lining of the microcirculation (Figure 1) [12][1]. Variations in cytokine concentrations at the site of inflammation, which develop over time, and different cytokines acting in concert within the cytokine network, are mechanisms by which the proinflammatory cytokines fine-tune the phases of acute inflammatory reaction, which evolves from rarefaction of the noxious stimulus, via activation of the specific immune response, to heal damaged tissue (Figure 1) [12][1]. During an acute inflammatory response, the cytokine IL-18 acts synergistically with other proinflammatory cytokines of the IL-1 family, using the same downstream signaling pathways to enhance production of these proinflammatory cytokines [5][13][72,83]. In the course of this pathway, IL-18 initiates the signaling cascades by binding to its heterodimeric receptor, which consists of the ligand-binding chain IL-18Rα and the signal-transduction chain IL-18Rβ. Upon ligation of IL-18 to the ligand-binding chain, these two parts of the receptor approximate, allowing for the trimeric complex to assemble, which is then capable of initiating downstream signaling [13][83]. The specific role of this cytokine is to induce the cytokine IFN-γ, whose role is to activate macrophages and generate the cytotoxic, cell-mediated immune response. In addition to macrophages, it involves NK and Th1 cells, too, and is essential for eliminating intracellular pathogens and aberrant and damaged cells (Figure 1) [14][15][84,85]. The signaling pathway engaged in producing IFN-γ takes the TRIF adaptor molecule-dependent pathway, which employs the endocytosed TLRs and induces IFN-γ synthesis via activation of the IFN-γ-inducing transcription factor IRF3 (Figure 1) [6][8][78,80]. Given the self-aggressive potential of cell-mediated immunity, strictly regulated IL-18-dependent signaling is essential for preventing uncontrolled tissue damage. In physiological conditions, however, the activity of the cytokine IL-18 is mainly regulated by IL-18BP, which binds this cytokine with higher affinity than the ligand-binding unit of the IL-18 receptor and thus suppresses the effect of this cytokine in generating the Th1-mediated immune response [14][15][84,85]. A rise in the production of the proinflammatory cytokines is an obligatory precondition for initiating the specific (T cell-mediated) immune response [15][85]. Namely, if present in the microenvironment as a sign of danger, the proinflammatory cytokines provide an activation signal to DCs by upregulating the costimulatory molecules and peptides of the major histocompatibility complex (MHC) on their surface. This step is necessary so that DCs can acquire the capability to prime naïve CD4+ Th lymphocytes to differentiate into one of the effector Th cell subsets, including Th1, Th2, and Th17 cell subsets (Figure 1) [15][85].
1.2. Suppression of Acute Inflammation by the Cytokine IL-37
The broad spectrum of suppressor activities of the cytokine IL-37 relies on the fact that this cytokine acts at strategic points in the proinflammatory pathways. Its mode of action primarily involves suppressing the effects of the proinflammatory cytokine IL-18, and thus, indirectly, also the effects of IL-1β [9][73]. The cytokine IL-37 induces its effects by binding to the IL-18Rα receptor component, which then attracts the decoy receptor IL-1R8 instead of the signal-conducting component IL-18Rβ (Figure 2). The newly developed IL-37/IL-18Rα/IL-1R8 receptor complex reduces IL-1β and IL-18-dependent signaling and can also reduce TLR-dependent signaling by sequestering the adaptor molecule MyD88. This results in dumping activities of the major intracellular proinflammatory pathways, including the IKK- and MAPK-dependent pathways, which are needed to control synthesis of the proinflammatory cytokines and to keep the PIK3/Akt/mTOR pathway active, whose role is to control the metabolic effects of inflammation (Figure 1) [9][16][73,86]. Although signaling pathways of the cytokine IL-37 have not been completely clarified, it seems that, in addition to dumping the proinflammatory pathways, IL-37 also acts to actively promote the anti-inflammatory pathways, operating via the negative signal molecules, such as STAT3, Mer, and PTEN (Figure 2) [9][17][73,87]. Apart from operating via the cell-surface receptors, IL-37 may also translocate to the nucleus, where it binds to the nuclear DNA and suppresses transcription of the proinflammatory genes (Figure 2). To be endowed for this nuclear activity, the IL-37 precursor molecules must be processed into the mature forms via proteolytic enzyme caspase-1 activity. The mature molecules can enter the nucleus after forming complexes with the molecule Smad 3—a kinase operating in the pathway of the immunosuppressive cytokine TGF-β (Figure 2).
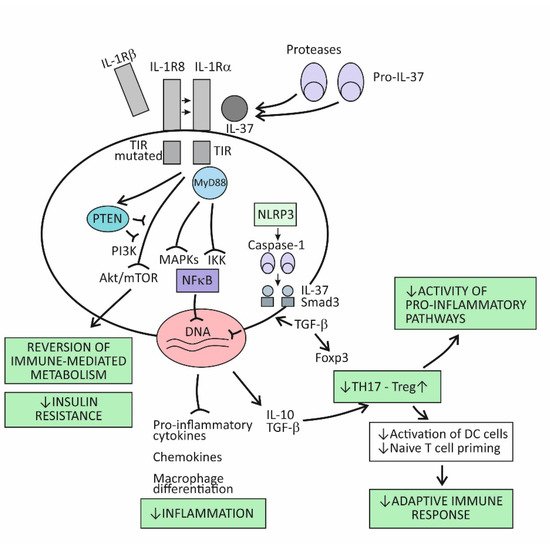
Figure 2.
The immunosuppressive and metabolic effects of the cytokine IL-37.
Emerging evidence indicates that IL-37 is constitutively expressed in human cells at low levels but is upregulated by inflammatory stimuli [9][16][17][73,86,87]. When intracellular concentrations of the mature form of this cytokine reaches some threshold, their precursor molecules are exported from the cell to the extracellular space. The extracellular proteases (for example, released from activated neutrophils) are thought to process the IL-37 precursors into the mature forms, which then may exert the extracellular functions (Figure 2). By modulating inflammation and the innate immune response, IL-37 also affects the adaptive (T cell-mediated) immune response. One of the proposed mechanisms is suppressing the stimulation of naïve Th lymphocytes by DCs, which is possible via reduced expression of costimulatory molecules such as CD40 and CD86 and MHC molecules on the surface of DCs. Another important mechanism is promoting the development of Treg cells, whose role is to limit inflammation and tissue damage by suppressing the generation of effector T-cell subsets (Figure 2) [18][88].
2. The Cytokine IL-37 as the Key Regulator of Chronic Inflammation Associated with Organ Damage in Age-Related Metabolic and Vascular Conditions
2.1. Immune-Metabolic Disturbances in Age-Related Metabolic and Vascular Conditions
As recent evidence suggests, inflammation and metabolism are inextricably linked at both the molecular and system levels and should not be explored separately [19][89]. Regarding cells of the immune system, their metabolic status is a critical determinant of their function [20][90]; on the other hand, while executing different functions in inflammation and host defense, immune cells adjust their metabolic pathways and usually need an additional supply of some nutrients in order to function and proliferate [21][91]. When viewed from the whole-body perspective, chronic inflammation has been recognized as an independent risk factor for the development and progression of age-related metabolic and vascular disorders, such as obesity, hypertension, T2D, and CVD [22][23][24][6,7,8]. Conversely, the presence of metabolic disorders, as we ahave already stated above, can accelerate aging via the release of a wide range of molecules that serve as danger signals and can become augmented [25][26][27][28][11,12,14,92]. In addition, intestinal dysbiosis usually accompanies obesity and T2D, representing a large source of microbial signals; in this way, and owing to insufficient synthesis of some essential nutrients, this condition may contribute to persisting inflammation [29][30][13,93]. A better understanding of mechanisms that operate at the crossroads between the innate immune system and metabolic disorders could enable us to develop improved therapies for curing chronic inflammatory and metabolic disorders. In this regard, it has been recognized that the same pro-inflammatory intracellular signaling pathways are used irrespective of the types of challenge, whether in response to infection or when driven by metabolic cues or increased tissue damage, as in the presence of chronic diseases [20][25][26][27][11,12,14,90]. In this context, inflammasomes play the key role through their effects in integrating proinflammatory signals and modulating metabolic pathways, mostly by controlling production of the proinflammatory cytokines (Figure 1) [31][94]. Accordingly, increased activity of the inflammasome NLRP3 has been found in metabolic and CV conditions [27][32][14,22]. Two distinct processes that operate at the crossroads of metabolic and pro-inflammatory pathways and that are of the utmost importance for the development and progression of age-related metabolic and vascular conditions deserve to be described in more detail. They include (1) trained immunity and (2) insulin resistance.
2.2. Trained Immunity
Until recently, immunological memory (an ability of the immune system to mount a more efficient defense reaction in repeated encounters with the same microorganism) was considered to be an exclusive feature of the adaptive (T cell-mediated) immune system [15][85]. A growing body of evidence, however, indicates that long-term adaptive changes may also affect monocytes/macrophages, resulting in their enhanced responses to repeated stimulation with infectious and noninfectious challenges [33][95]. As indicated by the results of transcriptional and epigenetic studies, those genes whose activity is affected in trained immunity are the ones involved in immune functions and in stabilizing glycolytic metabolic pathways [34][96]. Such studies have revealed the key mechanisms underlying trained immunity, including rewiring of cellular metabolism and induction of post-translational histone modifications (epigenetic changes). These mechanisms result in increased chromatin accessibility for inflammatory stimuli and a long-term increase in production of the proinflammatory cytokines. An essential step in the process of epigenetic reprogramming is the switch in cell energy metabolism from oxidative phosphorylation to glycolysis [21][35][36,91]. This process is regulated by activating the Akt/mTOR/Hif pathway, resulting in increased production of lactate and disruption of the tricarboxylic acid cycle (TCA), also known as the Krebs cycle (Figure 3). The purpose of these metabolic changes is to meet the requirement of activated immune cells to rapidly generate adenosine triphosphate (ATP), the energy-storage molecules needed to execute immune cell functions and synthesize new components [21][36][37][91,97,98]. If Krebs cycle activity decreases and some alternative metabolic pathways are activated, the intracellular concentrations of some metabolites increase, for example, citrate, succinate, and fumarate. Increased availability of these metabolites within the cell were found to accelerate histone modifications and the development of epigenetic changes (Figure 3) [21][35][36,91]. Apart from the effects of epigenetic reprogramming, changes in intracellular concentrations of some metabolites, by-products of glycolytic metabolic pathway activation, especially including increased concentrations of lipid moieties such as FFAs, cholesterol, and cholesterol derivatives or some amino acids, may also have a role in changing signal transduction processes via mechanisms such as changes in composition of the cell membrane and other cell structures or by directly influencing the signaling pathways [21][36][37][91,97,98].
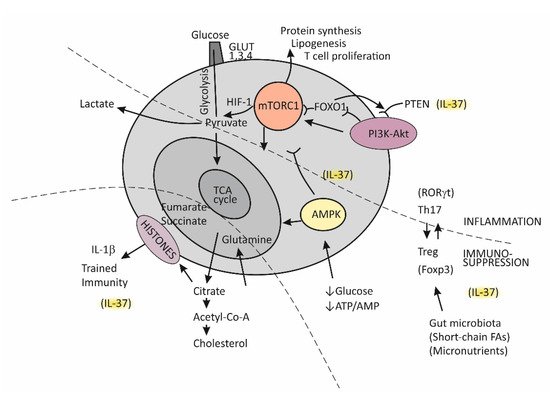
Figure 3.
Metabolic reprograming in chronic inflammation and the balance between the tolerogenic and inflammatory immune axis.
2.3. The Complex Molecule mTOR-Mediated Regulation of the Effector T-Cell Commitment
The serine threonine kinase, termed mammalian target of rapamycin (mTOR), has emerged as the key regulator of immune functions (Figure 3) [36][97]. The assembly of mTOR with different adapter proteins may form two different variants with different functional abilities. The variant mTORC1 has an essential role in committing naïve Th cells to differentiate into the effector Th1 and Th17 cell subsets, whereas mTORC2 signaling regulates differentiation of the Th2 cell subset [36][37][97,98]. The kinase mTORC1 is activated through signaling of the PI3K-Akt pathway and, in cells, has a role in regulating essential functions such as metabolism, protein synthesis, proliferation, and survival by sensing and integrating information on the availability of nutritional and growth factors. In T cells, activation of mTOR has a central role in regulating T-cell differentiation [37][98].
In general, when T cells are in the resting state, as is the case for naïve Th cells, they employ the catabolic mode of cellular metabolism, using autophagy to secure amino acids for protein synthesis and mitochondrial oxidative phosphorylation to maintain energy production [37][38][98,99]. This quiescent state is actively maintained, being controlled by regulatory transcription factors such as FOXO1, which was found to promote expression of inhibitory proteins, for example, PI3K inhibitor phosphatase and tensin homolog (PTEN) [39][100]. Upon activation, in contrast, T cells turn on the anabolic metabolic mode by switching from regular metabolism based on the Krebs cycle to prevalent participation in the cell metabolism of glycolysis; this switch is associated with increased nutrient uptake and biosynthetic activities. Activation of the complex mTORC1 is needed to sustain glycolytic metabolic reprograming [37][98]. The critical requirement, which regulates the activity of mTORC1, is sufficient supply of the cell with branched-chain amino acids, such as leucine and glutamine. An increase in the intracellular ATP/AMP ratio as a consequence of the switch of T cells to glycolytic metabolism suppresses activation of the AMP-kinase (AMPK), thus protecting mTORC1 from inhibitory activity of this kinase [37][40][98,101]. During T cell proliferation, the cell glucose transporters GLUT1, GLUT3, and GLUT4 are also induced and anchored to the cell membrane to promote glucose utilization [37][98].
It is important to mention that effector T cells, including cytolytic CD8+ T cells and Th1, Th2, and Th17 CD4+ effector cell subsets, are highly dependent on glycolytic metabolic reprogramming and that memory T cells and Treg cells use low levels of glucose and primarily utilize FFAs for oxidation and maintaining energy homeostasis (Figure 3) [37][38][98,99]. The activated PI3K-Akt signaling pathway upstream of mTORC1 increases the expression of glucose transporters, thus enhancing glycolysis and skewing the Th17/Treg balance [38][41][31,99].
A growing body of evidence indicates that Treg cells are equipped with some degree of plasticity, which is driven by oscillations in metabolic programming [38][39][41][31,99,100]. In the proliferation state, such as when Tregs recognize antigens and then migrate to the site of infection, Tregs regulate energy metabolism through the mTORC1-dependent pathway; otherwise, by turning on TORC1-independent metabolic pathways or oxidative phosphorylation, these cells regain a suppressor function. Taken together, mTORC1 signaling is necessary for Treg cells to proliferate and establish a suppressor function by promoting cholesterol/lipid metabolism. In the next steps of Treg cell generation, activation of the PIK3/Akt signaling pathway needs to be fine-tuned and well dosed in order to maintain Treg cell renewal and function.
In conditions when nutrients such as glucose or energy levels are low, AMPK activation, as the key cellular energy sensor, exerts an inhibitory effect on the activity of mTORC1, thus negatively regulating ATP-consuming biosynthetic processes and upregulating FFA oxidation and substance supply by autophagy (Figure 3) [36][40][41][31,97,101]. The production of IFN-γ is especially sensitive to glucose availability, and AMPK acts to suppress IFN-γ mRNA translation under conditions of glucose deprivation [40][101]. Proliferating T effector cells may eventually continue to upregulate the TCA cycle if there is a sufficient supply of amino acid glutamine to the cell by engaging glutamine to produce pyruvate, the main energy source for the TCA cycle [38][99]. By responding to a decreased amount of oxygen in the cell, the transcription factor Hif1 becomes activated and induces glycolytic enzymes, enabling promotion of Th17 cell differentiation [41][31]. Thus, the role of HifF-1 is to sustain the persistence/survival of Th17 cells in conditions associated with tissue hypoxia, such as in severely inflamed tissue [38][41][31,99]. The stability of the established Th17/Treg balance, under certain metabolic conditions, is maintained by epigenetically modifying RORγt and Foxp3 expression. Many specific metabolites that alter the tolerance/inflammation balance by upregulating either FOXP3 or RORγt expression have been identified [38][99]. A tolerogenic response is promoted, for example, when short-chain FFAs are processed by commensals in the colon [30][93]. This usually occurs in situations when an individual is in good health and in the absence of metabolic disturbances such as obesity or T2D [42][102].
2.4. Insulin Resistance
Under normal physiological conditions, insulin acts via insulin receptors (IRs) and stimulates glucose utilization to produce energy in insulin-dependent tissues (skeletal muscle, adipose tissue, the liver, pancreas, and the brain) [43][103]. The effects of insulin on metabolism extend to promoting glycogen and protein synthesis in muscle, lipogenesis in adipose tissue, and glycogen and FFA storage in the liver, via its inhibition effect for gluconeogenesis and glycogenolysis. In short, activation of IRs leads to tyrosine phosphorylation of insulin receptor substrate-1 (IRS-1), which, via activation of the PI3K-Akt signaling pathway, mediates glucose transport and other insulin-dependent effects. An intact PI3K-Akt signaling pathway is therefore critical for maintaining tissue homeostasis, as it mediates vital cellular processes, such as oxygen-dependent energy production, growth, proliferation, and survival. Impairments in this pathway induce insulin resistance (reduced tissue responsiveness to the physiological action of insulin), obesity, and T2D in insulin-sensitive tissues [44][45][104,105]. Conversely, metabolic disturbances associated with obesity often exacerbate insulin resistance by modulating this pathway, which, via excessive insulin production in the pancreatic β-cells, leads to a vicious circle of glucose homeostasis impairment, ultimately resulting in hyperglycemia and the development of T2D [43][103].
The process of activating the PI3K-Akt pathway is initiated when activated PI3K phosphorylates its substrate phosphatidylinositol biphosphate in intracellular membranes, thereby recruiting signaling proteins such as Akt. In fact, Akt becomes fully activated through two phosphorylation processes, including that on the threonine residue and that on the serine residue. Once activated, Akt regulates processes such as translation of GLUT4 and cellular energy production by stimulating glycolysis. At the same time, Akt reduces gluconeogenesis and FFA oxidation by inhibiting the protein FoxO1 and glycogen synthase kinase 3 (GSK3) and reduces protein synthesis by inhibiting mTORC1, whereas it increases cholesterol and FFA accumulation by activating the sterol regulatory element-binding proteins (SREBPs). The molecule PTEN is known to be the main negative regulator of PIK3 [43][103].
Dysfunctions in the PI3K/Akt pathway, by increasing insulin resistance and impairing glucose transport and glycogen synthesis, play a crucial role in the development of obesity and T2D. In contrast, obesity- and T2D-related disorders, such as increased production of FFAs and intracellular lipid deposition, mitochondrial dysfunction and increased oxidative stress, and increased production of proinflammatory cytokines, may promote insulin resistance by acting at different points of the PI3K/Akt pathway, especially in skeletal muscle, where the majority of insulin-stimulated glucose utilization takes place [44][104]. These disorders may promote insulin resistance by activating stress kinases (instead of Akt kinases), such as the c-Jun N-terminal kinase (JNK) family of MAPK kinases, and IKK kinases, which act to inhibit insulin-mediated signal transductions by triggering the inhibitory serine phosphorylation of IRS-1.
When this process is viewed from a more global perspective, it can be said that obesity-related disorders create an inflammatory microenvironment associated with increased intracellular formation of inflammasome complexes, notably including the NLRP3 type of inflammasome and thus amplifying both the innate immune response as well as the immune response locally, in tissue, and at the system level [31][46][94,106]. A growing body of evidence indicates that activation of inflammasomes by DAMPs plays a crucial role in obesity-induced inflammation and development of insulin resistance [46][47][106,107]. Moreover, the well-established view today is that an inflammatory state induced by the excess of nutrients and metabolic disorders, termed metainflammation, together with the chronic low-grade inflammation that accompanies aging, termed inflammaging, is the key driving force in the course of developing chronic diseases and functional deficits in older individuals [48][108].
2.5. The Role of the Cytokine IL-37 in Reversing Immune-Metabolic Disturbances Associated with Chronic Inflammation
After reviewing the most important processes at the crossroads of metabolic disorders and chronic inflammation, the multiple roles of the cytokine IL-37 in reversing these disorders have become easier to understand. The dual role (extracellular and intracellular) of this cytokine in suppressing production of proinflammatory cytokines, mostly including IL-1β and IL-18, can explain its role as the major natural inhibitor of immune responses in chronic inflammatory and autoimmune diseases, including cancer [49][109]. Moreover, recent evidence indicates that the biological effects of this cytokine extend beyond direct suppression of proinflammatory cytokines and also involve its effects on cell metabolism. The most important metabolic effect of IL-37 is in activating the MAPK and suppressing the Akt/mTORC/HIF-1 signaling pathways, which may reverse the established bias from mainly glycolysis to predominantly the participation of oxidative phosphorylation in cell metabolism [18][49][88,109].
Closely related to its effect in modulating the metabolic pathways, this cytokine is also involved in reversing the effects of the aberrant activation of trained immunity, as is the case in chronic inflammatory conditions (Figure 2) [49][109]. This effect of IL-37 was confirmed in a number of experimental models. It has been shown, for example, that treating experimental animals with IL-37 may reduce the expression of epigenetic markers of trained immunity or reverse metabolic changes, characteristics of trained immunity. In studies using human cell cultures extracted from individuals suffering from chronic health conditions, IL-37 was shown to inhibit inflammatory mediator production. In aged experimental animals, transgenic expression of IL-37 was shown to restore the function of both CD4+ and CD8+ T lymphocytes, which indicates its role in reversing the effects of chronic inflammation on age-related decline in immune responses [50][110]. Treatment of experimental animals with human recombinant IL-37 was found to reverse the metabolic costs that are often incurred in systemic inflammation and improve oxidative respiration and exercise tolerance in experimental animals [51][111]. The metabolic effects of IL-37, such as the effect on improving sensitivity to therapy with insulin in patients with T2D, could be, even partly, caused by the effect of IL-37 in reversing intestinal dysbiosis [52][112]. In a series of experiments, IL-37 has also been shown to play a pivotal role in fibrotic processes, associated with tissue and organ remodeling. Actually, the results of these experiments indicate that the lack of IL-37, either for genetic reasons or due to inappropriate upregulation in tissues as an expression of failed homeostatic regulatory mechanisms, is associated with increased tissue fibrosis [53][54][113,114]. Thus, in patients with idiopathic pulmonary fibrosis, a progressive and destructive lung disorder of unknown origin, the amount of IL-37 was found to be lower in alveolar epithelial cells and alveolar macrophages than in healthy controls [54][114]. As shown by in vitro experiments, mechanisms by which IL-37 may attenuate fibrotic processes are probably manifold, including decreased expression of mRNA and fibronectin and collagen synthesis in fibroblasts, inhibition of fibroblast proliferation on stimulation with TGF-β, and improved autophagy in fibroblasts. As indicated by experimental studies, variations in IL-37 concentrations can modulate inflammatory signals and the magnitude of tissue inflammatory responses to environmental challenges by being positioned at strategic places, such as in alveolar or intestinal epithelial cells and alveolar macrophages, that is, at the interface of the mucosal immune system and the outer environment [55][115].