Vitamin D is a semicosteroid hormone. It has beneficial effects on several bodyronutrient that is metabolised into a multifunctional secosteroid compound, calcitriol [1,25(OH)2D], esystems other than the musculoskeletal systemsential for the health and survival of humans. Both 25 dihydroxy vitamin D [25(OH)2D] and its active hormonal form, 1,25-dihydroxyvitamin D [1,25(OH)2D] are essential for human physiological functions, including damping down inflammation and the excessive intracellular oxidative stresses. Vitamin D is one of the key controllers of systemic inflammation, oxidative stress and mitochondrial respiratory function, and thus, the aging process in humans. In turn, molecular and cellular actions form 1,25(OH)2D slow down oxidative stress, cell and tissue damage, and the aging process. On the other hand, hypovitaminosis D impairs mitochondrial functions, and enhances oxidative stress and systemic inflammation. The interaction of 1,25(OH)2D with its intracellular receptors modulates vitamin D–dependent gene transcription and activation of vitamin D-responsive elements, which triggers multiple second messenger systems.
- 25(OH)D
- aging
- cytokines
- inflammation
- morbidity and mortality
- prevention
- reactive oxygen species
- ultraviolet
1. Introduction
Vitamin D is a micronutrient that is metabolised into a multifunctional secosteroid compound, calcitriol [1,25(OH)2D], essential for the health and survival of humans. Vitamin D deficiency has become a global pandemic, a significant public health problem affecting all ages and ethnic groups. It has surpassed iron deficiency as the most common micronutrient deficiency in the world. The prevalence of vitamin D deficiency continued to increase with associated complications, especially in countries furthest from the equator.
However, the incidence is also high among those who live within 1,000 km of the equator (e.g., in Sri Lanka, India, and Far Eastern, Middle Eastern, Central American, and Persian Gulf countries), primarily because of sun avoidance behaviour, due to harsh climatic conditions and desire to prevent skin becoming darker following sun exposure. Ethnic and cultural habits and having darker skin colour contributes to hypovitaminosis D [1][2][3][4].
Most of the vitamin D needed for humans is supposed to be generated from sun exposure (to summer-like sunlight, at least a third of the skin surface is exposed between 10.30 AM and 1.30 PM for up to an hour daily basis. The amount of D3 is generated dependent on the skin colour, time of the day, and location. In contrast, dietary sources play a supporting role when sunlight exposure is limited, or skin is ineffective in vitamin D production. Despite the above, more than 50% of the population has vitamin D deficiency at all times or sometime during the year [5][6]. If practical public health guidelines are implemented, vitamin D deficiency can be efficiently and cost-effectively treated and prevented. This approach will save lives and millions of dollars on vitamin D deficiency-associated disorders. While excessive sun exposure does not cause hypervitaminosis D, it can cause dermal cell DNA damage [7][8][9]. Thus, each country must develop safe sun exposure guidelines for its citizens.
2. Vitamin D and Gene Regulation
1,25(OH)2D regulates over 1,000 essential genes in the human genome [10]. The circulating 25(OH)D concentration is a helpful guide to vitamin D status. However, intracellularly generated calcitriol in target tissue cells, gene polymorphisms of its receptor, and the status of other micronutrients also influence the effectiveness and modulating of its autocrine/paracrine and second messenger signalling. Besides, epigenetic changes also modulate the ability of the calcitriol-bound VDR to bind to vitamin D response elements (VDREs) on promoter regions in genes and initiate second messenger systems [11]. The National Human Genome Research Institute (NHGRI) has launched a public research consortium, ENCODE [Encyclopedia of DNA Elements; http://www.genome.gov/10005107], to answer pertinent questions [12].
ENCODE has demonstrated the genome-wide actions of 1,25(OH)2D3 on the formation rates of proteins, such as the insulator protein CTCF (transcriptional repressor CTCF; 11-zinc finger protein, CCCTC-binding factor, etc.) and the VDR [13][14]. These findings suggest the presence of numerous functional VDRE regions across the human genome [11][15]. In addition, it has been suggested that the expression of these genes could be used as biomarkers for different actions of vitamin D in various tissues and cells and for assessing vulnerabilities [16].
Calcitriol also modulates growth and cell proliferation through direct and indirect pathways. For example, vitamin D inhibits the pathways related to the transcription factor NF-κB [17], which is crucial for many actions of the CD4 and other immune cells. People with chronic non-communicable diseases, such as cardiovascular disease, type 2 diabetes, autoimmune diseases, arthritis, and osteoporosis reported having chronically elevated NF-κB [18], secondary to chronic hypovitaminosis D. NF-κB enhances the oxidative stress and cellular responses to inflammation and injury, including following head injury [19]. Whereas calcitriol suppresses NF-κB and thereby reduces chronic somatic inflammation [20][21]. In addition, calcitriol also strengthens gap junctions, stabilises cell membranes, reduces cell proliferation and enhances cell differentiation—essential anti-cancer effects of vitamin D [22].
3. Vitamin D–Oxidative Stress
1,25(OH)2D is involved in many intracellular genomic activities and biochemical and enzymatic reactions, whereas D3 and 25(OH)D concentrations are crucial to diffuse these compounds into immune and other target cells. For example, calcitriol subdues the renin-angiotensin-aldosterone hormonal (RAS) axis. It stimulates the immune system to overcome inflammation and oxidative stress, enabling the destruction of pathogenic microbes, minimising cell damage from oxidative stress secondary to day-to-day exposure to toxic agents, and controlling the ageing process [23][24].
Physiologic circulating 25(OH)D concentration (i.e., between 50 and 80 ng/mL) enhances the expression of the nuclear factor, erythroid-2(Nf-E2)-related factor 2(Nrf2) [25][26][27] and also enhances Klotho, a phosphate regulating hormone and also an antiaging protein [28][29]. It also facilitates protein stabilisation [30]. Klotho also regulates cellular signalling systems, including forming antioxidants [31]. Consequently, in mice, functional abnormalities of the Klotho gene or removal of it through gene knock-out procedures induce premature ageing syndrome [32]. In animal studies, inefficient FGF23 and/or Klotho expression have been shown to cause premature ageing. Figure 1 is a schematic representation of various factors and their interactions that influence ageing and death.
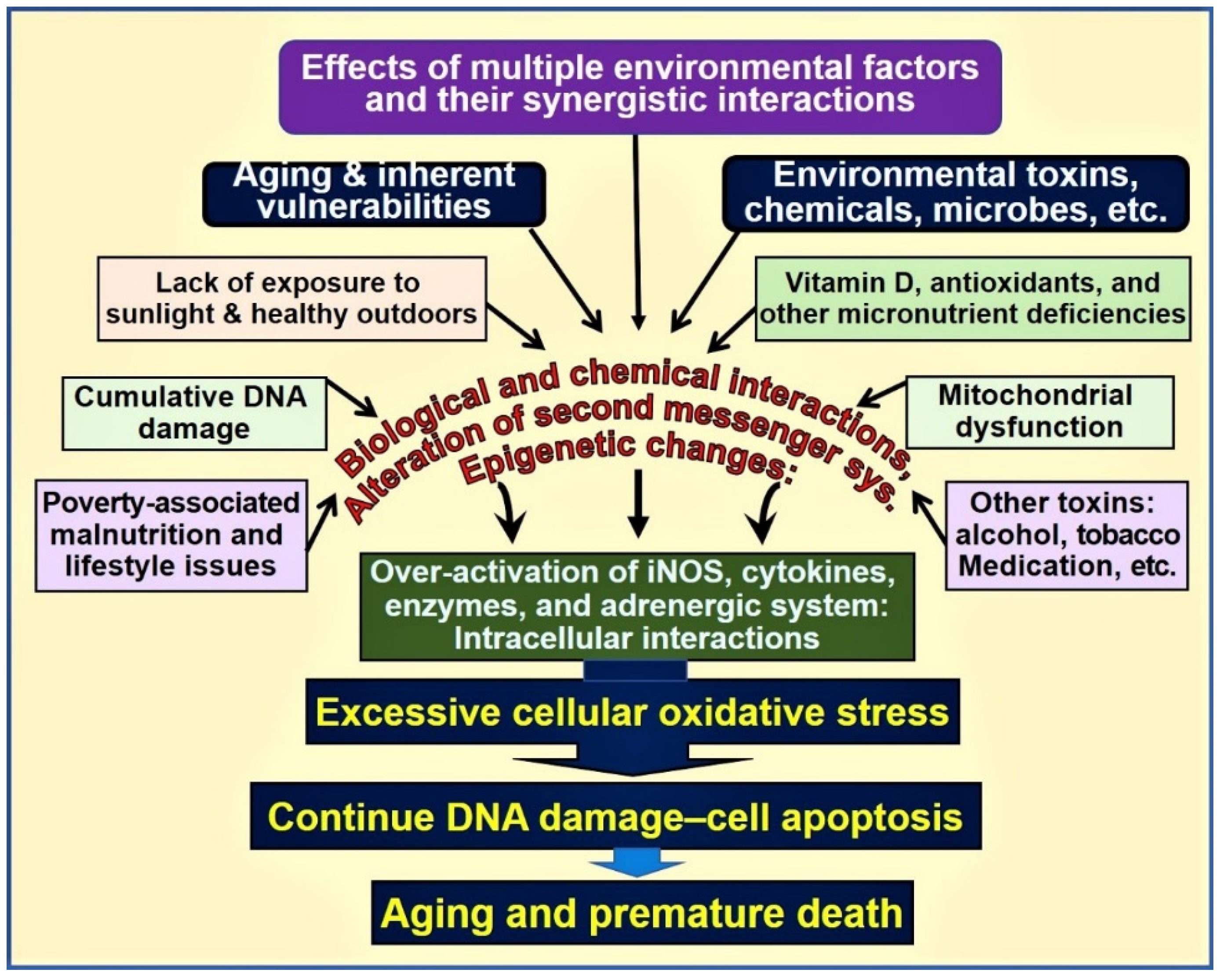
Figure 1. Environmental, microbial, biological, and chemical interactions modify the DNA and mitochondrial functions and epigenetics, modifying the ageing process. Vitamin D deficiency is one of the factors that enhances this oxidative-stress cycle and accelerates premature cell death [abbreviations used: DNA = deoxyribonucleic acid; iNOS = inducible nitric oxide enzyme].
3.1. Influences of Vitamin D on Oxidative Stress
When vitamin D is adequate, harmful intracellular oxidative stress-related activities are downregulated. Suboptimal concentrations of circulating 25(OH)D enhances oxidative stress and augments intracellular oxidative damage, including DNA and the rate of apoptosis. The intracellular Nrf2 level is inversely correlated with the accumulation of mitochondrial ROS [23][33] and the consequent escalation of oxidative stress. Thus, Nrf2 plays a key role in protecting cells against oxidative stress, modulated by vitamin D [34][35].
In addition, vitamin D supports cellular oxidation and reduction (redox) control by maintaining normal mitochondrial functions [36][37][38]. Loss in the redox control of the cell cycle may lead to aberrant cell proliferation, cell death, the development of neurodegenerative diseases, and accelerated ageing [38][39][40][41]. Peroxisome proliferator-activated receptor-coactivator 1α (PGC-1α) is bound to mitochondrial deacetylase (SIRT3). PGC-1α directly couples to the oxidative stress cycle [42] and interacts with Nrf2. This complex regulates the expression of SIRT3; this process is influenced by vitamin D metabolites [43]. In addition, the activation of the mitochondrial Nrf2/PGC-1α-SIRT3 path is dependent on intracellular calcitriol concentrations.
Calcitriol has overarching beneficial effects in upregulating the expression of antioxidants and anti-inflammatory cytokines [44], consequently protecting the tissues from toxins, micronutrient deficiency-related abnormalities, and parasitic (helminths) and intracellular microbe-induced harm [45]. It regulates ROS levels through its anti-inflammatory effects and mitochondrial-based expression of antioxidants through cell-signalling pathways [40][46].
4. Role of Vitamin D in Neutralisation of Toxins and Aging-Related Compounds
4.1. The Concept and the Process of Aging
Ageing generally refers to the biological process of growing older, also known as cellular senescence, a complex process. Advancing age, beginning from adulthood, is associated with a gradual decline of homeostatic mechanisms in maintaining health as described above, physiological functions and the capacity for regeneration [47]. Ageing has also been quantified from mortality curves using mathematical modelling; for example, by using the Gompertz equation m(t) = AeGt, for which m(t) = the mortality rate as a function of time or age (t); A = extrapolated constant to birth or maturity; G = the exponential (Gompertz) mortality rate coefficient] [48]. Many life insurance companies have used such to assess health risks.
Moreover, efficiency and the functions of the body decline after sexual maturity, suggesting a connection between the ageing process after fulfilling the procreation needs in vertebrates, including humans. Most age-related functions are irreversible, partly due to the accumulation of oxidative stress-related toxic products, methylation of DNA, and mitochondrial damage, and the inability to repair these wholly and efficiently, leading to reduced viability of cells and consequent accelerated cell death [49].
There is also a parallel decline in the immune system functions (i.e., immune-senescence) and an increase in systemic inflammation, demonstrable with an age-related increase of circulating pro-inflammatory cytokines [50][51]. These are likely to contribute to many age-related disorders, such as Alzheimer’s disease, cardiovascular, renal and pulmonary diseases, and susceptibility to autoimmunity and infections, especially the SARS-CoV-2 virus [50][51].
Many bodily functions slow with ageing, including response and reaction time; access to and the memory capacity; pulmonary, gastrointestinal, and cardiovascular capacities; and even the ability to naturally generate vitamin D in the skin. While age is perhaps the most potent risk factor for death, age-related disorders are the number one cause of death among older adults. The presence of vitamin D deficiency significantly aggravates this scenario.
Chronic hypovitaminosis D is associated with cardiovascular and metabolic dysfunctions and premature deaths [52], even among children [53]. In children, severe hypovitaminosis D increase Kawasaki-like syndrome and multi-system inflammatory disease following severe infections like COVID-19. Overall data suggest that vitamin D deficiency is important comorbidity, a risk factor for infections and premature death (i.e., all-cause mortality) [52][53][54][55]. Strong inverse relationships have been reported with vitamin D status with all-cause mortality [56][57][58], and cancer [58][59][60][61], etc.
4.2. Effects of Vitamin D on Apoptosis and Aging
The generalised chronic inflammatory and oxidative processes are known to cause cellular and DNA damage and increase apoptosis [62], as in the case of interstitial tubular cell damage in chronic kidney disease, and thus contribute to the ageing process [24][27][63]. In addition, hypovitaminosis D and dysfunctional mitochondrial activity significantly increase inflammation [46][64][65]. Thus, the anti-inflammatory effects of having physiological vitamin D concentrations (> 50 ng/mL) are crucial for better health and survival [63][66]. As mentioned above, hypovitaminosis D increases the expression of inflammatory cytokines [44][67], including tumour necrosis factor-α (TNF-α) and many other cytokines, increasing the expression of the InsP3Rs and resulting in increased intracellular Ca2+, causing accelerated cellular damage, apoptosis and ageing [39][68].
Multiple functions of the Klotho–Nrf2 regulatory system and their genes are regulated by calcitriol [29][35][38]. These include increasing intracellular antioxidant concentration and maintaining the redox homeostasis and a typical intracellular-reduced environment by removing excess ROS, thereby down-regulating the oxidative stress [69]. In addition, the vitamin D-dependent expression of γ-glutamyl transpeptidase, glutamate-cysteine ligase, and glutathione reductase contributes to the synthesis of the key redox agent glutathione (an essential antioxidant of low–molecular-weight thiol) [67][70]. In contrast, severe adverse outcomes occur in hypovitaminosis D.
Vitamin D also upregulates the expression of glutathione peroxidase that converts the ROS molecule H2O2 to water [70]. Vitamin D affects the formation of glutathione through activation of the enzyme glucose-6-phosphate dehydrogenase [70]—which downregulates nitrogen oxide (NOx), a potent precursor for generating ROS that converts O2− to H2O2 and upregulating superoxide dismutase (SOD). These vitamin D-related actions collectively reduce the burden of intracellular ROS.
Telomeres are repetitive DNA sequences that cap the ends of linear chromosomes protecting DNA molecules [71]. Ageing is associated with the shortening of telomeres, including in stem cells. The amount of telomerase gradually becomes too short to maintain its protective effects on DNA during cell division, and thus cell apoptosis. While vitamin D deficiency increases inflammation and intracellular oxidative stress, it also enhances the rate of telomere shortening during cell proliferation, resulting in genomic instability [18].
4.3. Hypovitaminosis D Leads to Deranged Mitochondrial Respiration
Intracellularly generated calcitriol in peripheral target cells is essential for maintaining physiological respiratory chain activity in mitochondria, facilitating energy generation [72][73]. In addition, 25(OH)D regulates the expression of the uncoupling protein attached to the mitochondria’s inner membrane that regulates thermogenesis [74][75][76]. Chronic vitamin D deficiency reduces the capacity of mitochondrial respiration through modulating nuclear mRNA [77][78][79]. The latter also downregulates the expression of complex I of the electron transport chain and thus reduces the formation of adenosine triphosphate (ATP) [40][68], another mechanism that increases cancer risks. Consequently, a low level of electron transport chain increases ROS formation and oxidative stress, a common phenomenon following acute and chronic exposure to toxins and many chronic diseases and seen in ageing [39][80][81].
The accumulation of intracellular toxins and/or age-related products disrupts signalling pathways, including the G protein-coupled systems, caspases, mitochondria, and the death receptor-linked mechanisms, triggering cell apoptosis and premature cell death: a known phenomenon in ageing [82][83]. The process is aggravated by stimulating G proteins leading to activation of downstream pathways, including protein kinase A and C (PKA and PKC), phosphatidylinositol-3-kinase (PI3-kinase), Ca2+ and MAP kinase-dependent systems, tyrosine phosphorylation [68][83], and work additively, aiding cancer genesis [61], and accelerating the ageing process.
4.4. Calcitriol Protects Mitochondrial Functions
Toxins, chronic metabolic abnormalities, and ageing processes are also known to cause mitochondrial dysfunction [39][75][76][77][84]. Abnormal mitochondria produce suboptimal amounts of ATP while generating excess ROS, creating a vicious cycle of enhanced and persisting effects from excessive oxidative stress [75][76][85]. These events cause DNA damage (and impairment of DNA repair enzymes), premature cell death, and accelerated ageing [35][39]. Cumulating data suggest that mitochondrial dysfunction is likely fueled by sustained intracellular inflammation, as in the case of vitamin D deficiency [48][63][65][86].
Dysfunctional mitochondria also have reduced intracellular Ca2+ buffering capacity, resulting in increased (and fluctuating) intracellular Ca2+ levels, which are “cytotoxic” and contribute to the impairment of cell regeneration and the sustenance of several chronic diseases [77][84]. Sub-physiological concentrations of calcitriol enhance and maintain oxidative stress, autophagy, inflammation, mitochondrial dysfunction (low ATP generation), adverse epigenetic changes, DNA damage, intracellular Ca2+, and generation and signalling of ROS. Therefore, sustained, adequate serum 25(OH)D concentrations (i.e., above 50 ng/mL) allow target tissues to remain healthy and overcome toxic effects and destructive processes [41][44][46]. Multiple benefits of controlling excessive oxidative stress are illustrated in Figure 2.
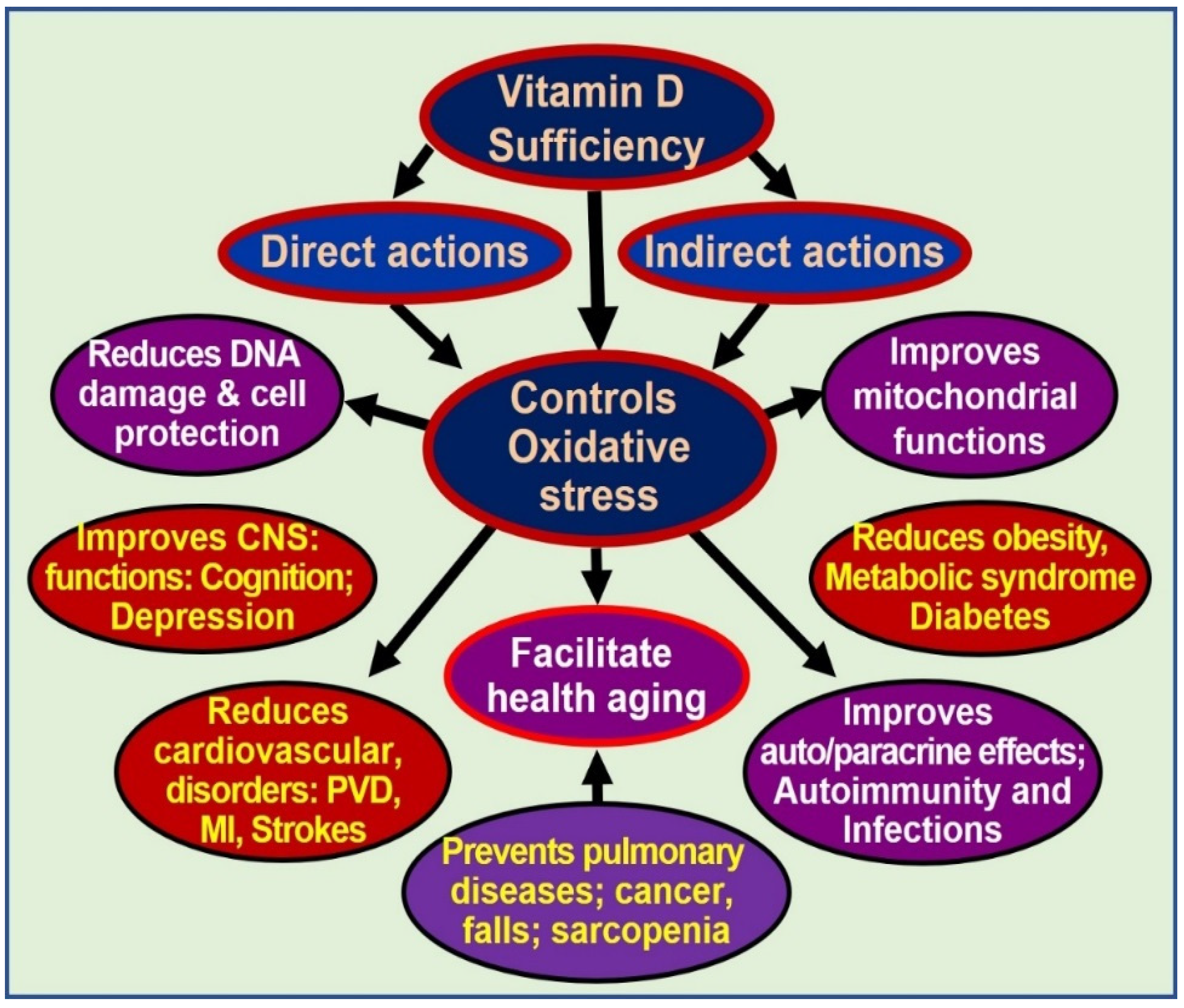
Figure 2. Oxidative stress is harmful to cells. Controlling oxidative stresses through vitamin D adequacy leads to cellular and organ protection and reduces ageing effects [abbreviations: CNS = central nervous system; DNA = deoxyribonucleic acid; MI = myocardial infarction; PVD = peripheral vascular diseases].
References
- Van Schoor, N.M.; Lips, P. Worldwide vitamin D status. Best Pract. Res. Clin. Endocrinol. Metab. 2011, 25, 671–680.
- Eggemoen, A.R.; Knutsen, K.V.; Dalen, I.; Jenum, A.K. Vitamin D status in recently arrived immigrants from Africa and Asia: A cross-sectional study from Norway of children, adolescents and adults. BMJ Open 2013, 3, e003293.
- Garland, C.F.; Gorham, E.D.; Mohr, S.B.; Garland, F.C. Vitamin D for cancer prevention: Global perspective. Ann. Epidemiol. 2009, 19, 468–483.
- Hilger, J.; Friedel, A.; Herr, R.; Rausch, T.; Roos, F.; Wahl, D.A.; Pierroz, D.D.; Weber, P.; Hoffmann, K. A systematic review of vitamin D status in populations worldwide. Br. J. Nutr. 2014, 111, 23–45.
- Haq, A.; Wimalawansa, S.J.; Carlberg, C. Highlights from the 5th International Conference on Vitamin D Deficiency, Nutrition and Human Health, Abu Dhabi, United Arab Emirates, March 24-25, 2016. J. Steroid Biochem. Mol. Biol. 2018, 175, 1–3.
- Pludowski, P.; Holick, M.F.; Grant, W.B.; Konstantynowicz, J.; Mascarenhas, M.R.; Haq, A.; Povoroznyuk, V.; Balatska, N.; Barbosa, A.P.; Karonova, T.; et al. Vitamin D supplementation guidelines. J. Steroid Biochem. Mol. Biol. 2018, 175, 125–135.
- Felton, S.J.; Cooke, M.S.; Kift, R.; Berry, J.L.; Webb, A.R.; Lam, P.M.; de Gruijl, F.R.; Vail, A.; Rhodes, L.E. Concurrent beneficial (vitamin D production) and hazardous (cutaneous DNA damage) impact of repeated low-level summer sunlight exposures. Br. J. Dermatol. 2016, 175, 1320–1328.
- Gordon-Thomson, C.; Tongkao-on, W.; Song, E.J.; Carter, S.E.; Dixon, K.M.; Mason, R.S. Protection from ultraviolet damage and photocarcinogenesis by vitamin D compounds. Adv. Exp. Med. Biol. 2014, 810, 303–328.
- Petersen, B.; Wulf, H.C.; Triguero-Mas, M.; Philipsen, P.A.; Thieden, E.; Olsen, P.; Heydenreich, J.; Dadvand, P.; Basagana, X.; Liljendahl, T.S.; et al. Sun and ski holidays improve vitamin D status, but are associated with high levels of DNA damage. J. Investig. Dermatol. 2014, 134, 2806–2813.
- Jeon, S.M.; Shin, E.A. Exploring vitamin D metabolism and function in cancer. Exp. Mol. Med. 2018, 50, 20.
- Valdivielso, J.M. The physiology of vitamin D receptor activation. Contrib. Nephrol. 2009, 163, 206–212.
- Farnham, P.J. Thematic minireview series on results from the ENCODE Project: Integrative global analyses of regulatory regions in the human genome. J. Biol. Chem. 2012, 287, 30885–30887.
- Washington, S.D.; Edenfield, S.I.; Lieux, C.; Watson, Z.L.; Taasan, S.M.; Dhummakupt, A.; Bloom, D.C.; Neumann, D.M. Depletion of the insulator protein CTCF results in HSV-1 reactivation in vivo. J. Virol. 2018.
- MacPherson, M.J.; Sadowski, P.D. The CTCF insulator protein forms an unusual DNA structure. BMC Mol. Biol. 2010, 11, 101.
- Carlberg, C. Genome-wide (over)view on the actions of vitamin D. Front. Physiol. 2014, 5, 167.
- Narvaez, C.J.; Matthews, D.; LaPorta, E.; Simmons, K.M.; Beaudin, S.; Welsh, J. The impact of vitamin D in breast cancer: Genomics, pathways, metabolism. Front. Physiol. 2014, 5, 213.
- Tilstra, J.S.; Robinson, A.R.; Wang, J.; Gregg, S.Q.; Clauson, C.L.; Reay, D.P.; Nasto, L.A.; St Croix, C.M.; Usas, A.; Vo, N.; et al. NF-kappaB inhibition delays DNA damage-induced senescence and aging in mice. J. Clin. Investig. 2012, 122, 2601–2612.
- Pusceddu, I.; Farrell, C.J.; Di Pierro, A.M.; Jani, E.; Herrmann, W.; Herrmann, M. The role of telomeres and vitamin D in cellular aging and age-related diseases. Clin. Chem. Lab. Med. 2015, 53, 1661–1678.
- Tang, H.; Hua, F.; Wang, J.; Sayeed, I.; Wang, X.; Chen, Z.; Yousuf, S.; Atif, F.; Stein, D.G. Progesterone and vitamin D: Improvement after traumatic brain injury in middle-aged rats. Horm. Behav. 2013, 64, 527–538.
- Wang, Q.; He, Y.; Shen, Y.; Zhang, Q.; Chen, D.; Zuo, C.; Qin, J.; Wang, H.; Wang, J.; Yu, Y. Vitamin D inhibits COX-2 expression and inflammatory response by targeting thioesterase superfamily member 4. J. Biol. Chem. 2014, 289, 11681–11694.
- Myszka, M.; Klinger, M. The immunomodulatory role of Vitamin D. Postepy Hig. Med. Dosw. 2014, 68, 865–878.
- Watanabe, R.; Inoue, D. Current Topics on Vitamin D. Anti-cancer effects of vitamin D. Clin. Calcium 2015, 25, 373–380.
- Holmes, S.; Abbassi, B.; Su, C.; Singh, M.; Cunningham, R.L. Oxidative stress defines the neuroprotective or neurotoxic properties of androgens in immortalized female rat dopaminergic neuronal cells. Endocrinology 2013, 154, 4281–4292.
- Petersen, K.S.; Smith, C. Ageing-Associated Oxidative Stress and Inflammation Are Alleviated by Products from Grapes. Oxid. Med. Cell. Longev. 2016, 2016, 6236309.
- Nakai, K.; Fujii, H.; Kono, K.; Goto, S.; Kitazawa, R.; Kitazawa, S.; Hirata, M.; Shinohara, M.; Fukagawa, M.; Nishi, S. Vitamin D activates the Nrf2-Keap1 antioxidant pathway and ameliorates nephropathy in diabetic rats. Am. J. Hypertens. 2014, 27, 586–595.
- Lewis, K.N.; Mele, J.; Hayes, J.D.; Buffenstein, R. Nrf2, a guardian of healthspan and gatekeeper of species longevity. Integr. Comp. Biol. 2010, 50, 829–843.
- Tullet, J.M.A.; Green, J.W.; Au, C.; Benedetto, A.; Thompson, M.A.; Clark, E.; Gilliat, A.F.; Young, A.; Schmeisser, K.; Gems, D. The SKN-1/Nrf2 transcription factor can protect against oxidative stress and increase lifespan in C. elegans by distinct mechanisms. Aging Cell 2017, 16, 1191–1194.
- Forster, R.E.; Jurutka, P.W.; Hsieh, J.C.; Haussler, C.A.; Lowmiller, C.L.; Kaneko, I.; Haussler, M.R.; Kerr Whitfield, G. Vitamin D receptor controls expression of the anti-aging klotho gene in mouse and human renal cells. Biochem. Biophys. Res. Commun. 2011, 414, 557–562.
- Berridge, M.J. Vitamin D: A custodian of cell signalling stability in health and disease. Biochem. Soc. Trans. 2015, 43, 349–358.
- Mark, K.A.; Dumas, K.J.; Bhaumik, D.; Schilling, B.; Davis, S.; Oron, T.R.; Sorensen, D.J.; Lucanic, M.; Brem, R.B.; Melov, S.; et al. Vitamin D Promotes Protein Homeostasis and Longevity via the Stress Response Pathway Genes skn-1, ire-1, and xbp-1. Cell Rep. 2016, 17, 1227–1237.
- Razzaque, M.S. FGF23, klotho and vitamin D interactions: What have we learned from in vivo mouse genetics studies? Adv. Exp. Med. Biol. 2012, 728, 84–91.
- Kuro-o, M. Klotho and aging. Biochim. Biophys. Acta 2009, 1790, 1049–1058.
- Tseng, A.H.; Shieh, S.S.; Wang, D.L. SIRT3 deacetylates FOXO3 to protect mitochondria against oxidative damage. Free Radic. Biol. Med. 2013, 63, 222–234.
- Wang, L.; Lewis, T.; Zhang, Y.L.; Khodier, C.; Magesh, S.; Chen, L.; Inoyama, D.; Chen, Y.; Zhen, J.; Hu, L.; et al. The identification and characterization of non-reactive inhibitor of Keap1-Nrf2 interaction through HTS using a fluorescence polarization assay. In Probe Reports from the NIH Molecular Libraries Program; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2010.
- Berridge, M.J. Vitamin D deficiency: Infertility and neurodevelopmental diseases (attention deficit hyperactivity disorder, autism, and schizophrenia). Am. J. Physiol. Cell Physiol. 2018, 314, C135–C151.
- Ryan, Z.C.; Craig, T.A.; Folmes, C.D.; Wang, X.; Lanza, I.R.; Schaible, N.S.; Salisbury, J.L.; Nair, K.S.; Terzic, A.; Sieck, G.C.; et al. 1alpha,25-Dihydroxyvitamin D3 Regulates Mitochondrial Oxygen Consumption and Dynamics in Human Skeletal Muscle Cells. J. Biol. Chem. 2016, 291, 1514–1528.
- Bouillon, R.; Verstuyf, A. Vitamin D, mitochondria, and muscle. J. Clin. Endocrinol. Metab. 2013, 98, 961–963.
- Sarsour, E.H.; Kumar, M.G.; Chaudhuri, L.; Kalen, A.L.; Goswami, P.C. Redox control of the cell cycle in health and disease. Antioxid. Redox Signal. 2009, 11, 2985–3011.
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795.
- Berridge, M.J. Vitamin D cell signalling in health and disease. Biochem. Biophys. Res. Commun. 2015, 460, 53–71.
- Ureshino, R.P.; Rocha, K.K.; Lopes, G.S.; Bincoletto, C.; Smaili, S.S. Calcium signaling alterations, oxidative stress, and autophagy in aging. Antioxid. Redox Signal. 2014, 21, 123–137.
- Chen, Y.; Zhang, J.; Lin, Y.; Lei, Q.; Guan, K.L.; Zhao, S.; Xiong, Y. Tumour suppressor SIRT3 deacetylates and activates manganese superoxide dismutase to scavenge ROS. EMBO Rep. 2011, 12, 534–541.
- Song, C.; Fu, B.; Zhang, J.; Zhao, J.; Yuan, M.; Peng, W.; Zhang, Y.; Wu, H. Sodium fluoride induces nephrotoxicity via oxidative stress-regulated mitochondrial SIRT3 signaling pathway. Sci. Rep. 2017, 7, 672.
- Wei, R.; Christakos, S. Mechanisms Underlying the Regulation of Innate and Adaptive Immunity by Vitamin D. Nutrients 2015, 7, 8251–8260.
- George, N.; Kumar, T.P.; Antony, S.; Jayanarayanan, S.; Paulose, C.S. Effect of vitamin D3 in reducing metabolic and oxidative stress in the liver of streptozotocin-induced diabetic rats. Br. J. Nutr. 2012, 108, 1410–1418.
- Shelton, R.C.; Claiborne, J.; Sidoryk-Wegrzynowicz, M.; Reddy, R.; Aschner, M.; Lewis, D.A.; Mirnics, K. Altered expression of genes involved in inflammation and apoptosis in frontal cortex in major depression. Mol. Psychiatry 2011, 16, 751–762.
- Macedo, J.C.; Vaz, S.; Logarinho, E. Mitotic Dysfunction Associated with Aging Hallmarks. Adv. Exp. Med. Biol. 2017, 1002, 153–188.
- Finch, C.E.; Pike, M.C. Maximum life span predictions from the Gompertz mortality model. J. Gerontol. A Biol. Sci. Med. Sci. 1996, 51, B183–B194.
- Jallali, N.; Ridha, H.; Thrasivoulou, C.; Underwood, C.; Butler, P.E.; Cowen, T. Vulnerability to ROS-induced cell death in ageing articular cartilage: The role of antioxidant enzyme activity. Osteoarthr. Cartil. 2005, 13, 614–622.
- Fulop, T.; Larbi, A.; Dupuis, G.; Le Page, A.; Frost, E.H.; Cohen, A.A.; Witkowski, J.M.; Franceschi, C. Immunosenescence and Inflamm-Aging As Two Sides of the Same Coin: Friends or Foes? Front. Immunol. 2017, 8, 1960.
- Franceschi, C.; Bonafe, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254.
- Pilz, S.; Marz, W.; Wellnitz, B.; Seelhorst, U.; Fahrleitner-Pammer, A.; Dimai, H.P.; Boehm, B.O.; Dobnig, H. Association of vitamin D deficiency with heart failure and sudden cardiac death in a large cross-sectional study of patients referred for coronary angiography. J. Clin. Endocrinol. Metab. 2008, 93, 3927–3935.
- Cohen, M.C.; Offiah, A.; Sprigg, A.; Al-Adnani, M. Vitamin D deficiency and sudden unexpected death in infancy and childhood: A cohort study. Pediatr. Dev. Pathol. 2013, 16, 292–300.
- Grant, W.B. Vitamin D Deficiency May Explain Comorbidity as an Independent Risk Factor for Death Associated with Cancer in Taiwan. Asia Pac. J. Public Health 2015, 27, 572–573.
- Scorza, F.A.; Albuquerque, M.; Arida, R.M.; Terra, V.C.; Machado, H.R.; Cavalheiro, E.A. Benefits of sunlight: Vitamin D deficiency might increase the risk of sudden unexpected death in epilepsy. Med. Hypotheses 2010, 74, 158–161.
- Autier, P.; Mullie, P.; Macacu, A.; Dragomir, M.; Boniol, M.; Coppens, K.; Pizot, C.; Boniol, M. Effect of vitamin D supplementation on non-skeletal disorders: A systematic review of meta-analyses and randomised trials. Lancet Diabetes Endocrinol. 2017, 5, 986–1004.
- Brenner, H.; Jansen, L.; Saum, K.U.; Holleczek, B.; Schottker, B. Vitamin D Supplementation Trials Aimed at Reducing Mortality Have Much Higher Power When Focusing on People with Low Serum 25-Hydroxyvitamin D Concentrations. J. Nutr. 2017, 147, 1325–1333.
- Autier, P.; Boniol, M.; Pizot, C.; Mullie, P. Vitamin D status and ill health: A systematic review. Lancet Diabetes Endocrinol. 2014, 2, 76–89.
- Bjelakovic, G.; Gluud, L.L.; Nikolova, D.; Whitfield, K.; Krstic, G.; Wetterslev, J.; Gluud, C. Vitamin D supplementation for prevention of cancer in adults. Cochrane Database Syst. Rev. 2014.
- Pilz, S.; Tomaschitz, A.; Marz, W.; Drechsler, C.; Ritz, E.; Zittermann, A.; Cavalier, E.; Pieber, T.R.; Lappe, J.M.; Grant, W.B.; et al. Vitamin D, cardiovascular disease and mortality. Clin. Endocrinol. 2011, 75, 575–584.
- Van der Reest, J.; Gottlieb, E. Anti-cancer effects of vitamin C revisited. Cell Res. 2016, 26, 269–270.
- Da Luz Dias, R.; Basso, B.; Donadio, M.V.F.; Pujol, F.V.; Bartrons, R.; Haute, G.V.; Gassen, R.B.; Bregolin, H.D.; Krause, G.; Viau, C.; et al. Leucine reduces the proliferation of MC3T3-E1 cells through DNA damage and cell senescence. Toxicol. In Vitro 2018, 48, 1–10.
- Cevenini, E.; Caruso, C.; Candore, G.; Capri, M.; Nuzzo, D.; Duro, G.; Rizzo, C.; Colonna-Romano, G.; Lio, D.; Di Carlo, D.; et al. Age-related inflammation: The contribution of different organs, tissues and systems. How to face it for therapeutic approaches. Curr. Pharm. Des. 2010, 16, 609–618.
- Talmor, Y.; Bernheim, J.; Klein, O.; Green, J.; Rashid, G. Calcitriol blunts pro-atherosclerotic parameters through NFkappaB and p38 in vitro. Eur. J. Clin. Investig. 2008, 38, 548–554.
- Morris, G.; Maes, M. Mitochondrial dysfunctions in myalgic encephalomyelitis/chronic fatigue syndrome explained by activated immuno-inflammatory, oxidative and nitrosative stress pathways. Metab. Brain Dis. 2014, 29, 19–36.
- Berk, M.; Williams, L.J.; Jacka, F.N.; O’Neil, A.; Pasco, J.A.; Moylan, S.; Allen, N.B.; Stuart, A.L.; Hayley, A.C.; Byrne, M.L.; et al. So depression is an inflammatory disease, but where does the inflammation come from? BMC Med. 2013, 11, 200.
- Beilfuss, J.; Berg, V.; Sneve, M.; Jorde, R.; Kamycheva, E. Effects of a 1-year supplementation with cholecalciferol on interleukin-6, tumor necrosis factor-alpha and insulin resistance in overweight and obese subjects. Cytokine 2012, 60, 870–874.
- Petersen, O.H.; Verkhratsky, A. Calcium and ATP control multiple vital functions. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2016, 371, 20150418.
- Calton, E.K.; Keane, K.N.; Soares, M.J.; Rowlands, J.; Newsholme, P. Prevailing vitamin D status influences mitochondrial and glycolytic bioenergetics in peripheral blood mononuclear cells obtained from adults. Redox Biol. 2016, 10, 243–250.
- Liu, Y.; Hyde, A.S.; Simpson, M.A.; Barycki, J.J. Emerging regulatory paradigms in glutathione metabolism. Adv. Cancer Res. 2014, 122, 69–101.
- Weipoltshammer, K.; Schofer, C.; Almeder, M.; Philimonenko, V.V.; Frei, K.; Wachtler, F.; Hozak, P. Intranuclear anchoring of repetitive DNA sequences: Centromeres, telomeres, and ribosomal DNA. J. Cell Biol. 1999, 147, 1409–1418.
- Consiglio, M.; Viano, M.; Casarin, S.; Castagnoli, C.; Pescarmona, G.; Silvagno, F. Mitochondrial and lipogenic effects of vitamin D on differentiating and proliferating human keratinocytes. Exp. Dermatol. 2015, 24, 748–753.
- Wyckelsma, V.L.; Levinger, I.; McKenna, M.J.; Formosa, L.E.; Ryan, M.T.; Petersen, A.C.; Anderson, M.J.; Murphy, R.M. Preservation of skeletal muscle mitochondrial content in older adults: Relationship between mitochondria, fibre type and high-intensity exercise training. J. Physiol. 2017, 595, 3345–3359.
- Constantinescu, A.A.; Abbas, M.; Kassem, M.; Gleizes, C.; Kreutter, G.; Schini-Kerth, V.; Mitrea, I.L.; Toti, F.; Kessler, L. Differential influence of tacrolimus and sirolimus on mitochondrial-dependent signaling for apoptosis in pancreatic cells. Mol. Cell. Biochem. 2016, 418, 91–102.
- Petrosillo, G.; Matera, M.; Casanova, G.; Ruggiero, F.M.; Paradies, G. Mitochondrial dysfunction in rat brain with aging Involvement of complex I, reactive oxygen species and cardiolipin. Neurochem. Int. 2008, 53, 126–131.
- Petrosillo, G.; Matera, M.; Moro, N.; Ruggiero, F.M.; Paradies, G. Mitochondrial complex I dysfunction in rat heart with aging: Critical role of reactive oxygen species and cardiolipin. Free Radic. Biol. Med. 2009, 46, 88–94.
- Yin, F.; Sancheti, H.; Patil, I.; Cadenas, E. Energy metabolism and inflammation in brain aging and Alzheimer’s disease. Free Radic. Biol. Med. 2016, 100, 108–122.
- Prior, S.; Kim, A.; Yoshihara, T.; Tobita, S.; Takeuchi, T.; Higuchi, M. Mitochondrial respiratory function induces endogenous hypoxia. PLoS ONE 2014, 9, e88911.
- Scaini, G.; Rezin, G.T.; Carvalho, A.F.; Streck, E.L.; Berk, M.; Quevedo, J. Mitochondrial dysfunction in bipolar disorder: Evidence, pathophysiology and translational implications. Neurosci. Biobehav. Rev. 2016, 68, 694–713.
- Zhu, Q.S.; Xia, L.; Mills, G.B.; Lowell, C.A.; Touw, I.P.; Corey, S.J. G-CSF induced reactive oxygen species involves Lyn-PI3-kinase-Akt and contributes to myeloid cell growth. Blood 2006, 107, 1847–1856.
- Lin, Y.; Berg, A.H.; Iyengar, P.; Lam, T.K.; Giacca, A.; Combs, T.P.; Rajala, M.W.; Du, X.; Rollman, B.; Li, W.; et al. The hyperglycemia-induced inflammatory response in adipocytes: The role of reactive oxygen species. J. Biol. Chem. 2005, 280, 4617–4626.
- Agalakova, A.A.; Gusev, G.P. Molecular mechanisms of cytotoxicity and apoptosis induced by inorganic fluoride. ISRN Cell Biol. 2012, 2012, 403835.
- Agalakova, N.I.; Gusev, G.P. Fluoride induces oxidative stress and ATP depletion in the rat erythrocytes in vitro. Environ. Toxicol. Pharmacol. 2012, 34, 334–337.
- Yin, F.; Sancheti, H.; Liu, Z.; Cadenas, E. Mitochondrial function in ageing: Coordination with signalling and transcriptional pathways. J. Physiol. 2016, 594, 2025–2042.
- Marzetti, E.; Calvani, R.; Cesari, M.; Buford, T.W.; Lorenzi, M.; Behnke, B.J.; Leeuwenburgh, C. Mitochondrial dysfunction and sarcopenia of aging: From signaling pathways to clinical trials. Int. J. Biochem. Cell Biol. 2013, 45, 2288–2301.
- Morris, G.; Berk, M. The many roads to mitochondrial dysfunction in neuroimmune and neuropsychiatric disorders. BMC Med. 2015, 13, 68.