Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is a comparison between Version 1 by Muhammad S Soyfoo and Version 2 by Lindsay Dong.
IL-33 is a newly discovered cytokine displaying pleiotropic localizations and functions. More specifically, it also functions as an alarmin, following its release from cells undergoing cell death or necrosis, to alert the innate immune system. The role of IL-33 has been underlined in several inflammatory and autoimmune diseases including systemic lupus erythematosus (SLE). The expressions of IL-33 as well as its receptor, ST2, are significantly upregulated in SLE patients and in patients with lupus nephritis.
- systemic lupus erythematosus
- IL-33
- cytokines
- autoimmune disease
1. Introduction
Systemic lupus erythematosus (SLE) is the prototype of autoimmune connective tissue disease and affects mainly young women of childbearing age [1]. It is characterized by chronic and aberrant immune activation against self-antigens, ultimately leading to the production of autoantibodies and immune complexes (ICs) entailing further damage in multiple organs such as the joints, skin, kidneys, lungs and brain [2]. Even though the contribution of the innate and adaptive immune systems to the break of tolerance towards autoantigens is well established, the exact mechanisms underlying this phenomenon still remain elusive.
IL-33 is a cytokine that was first identified approximately 20 years ago as a ligand for the IL-1 receptor (IL-1R) family member suppression of tumorigenicity 2 (ST2) [3][15], and it has been associated with several biological processes and plays a pivotal role in innate and adaptive immunity, tissue repair, homeostasis and responses to environmental stresses. IL-33 is believed to act as an alarmin, as it is passively released by damaged or necrotic barrier cells (endothelial and epithelial cells) [4][16]. Alarmins mediate intercellular signals through interactions with chemotactic and pattern recognition receptors (PRRs) to foster innate immune cells. Additionally, alarmins have the ability to elicit adaptive immunity responses and T cell-dependent long-term immune memory through their capacity to induce DC maturation [5][17]. IL-33 primarily induces type 2 helper (Th2) immune responses through its receptor ST2 [6][18]. However, recent studies found ST2 expression on Th1 cells, Treg cells, group 2 innate lymphoid cells (ILC2), CD8+ T cells and natural killer (NK) cells [7][8][19,20].
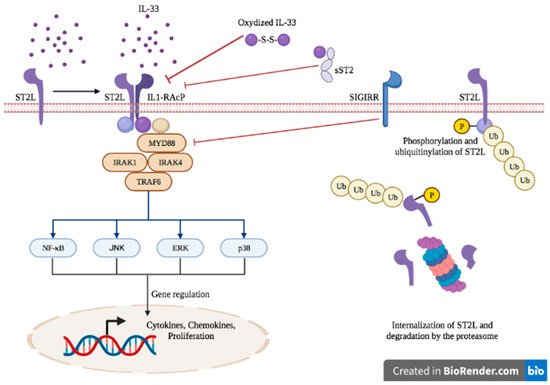
2. IL-33 and ST2: Biology and Functions
IL-33 is a member of the IL-1 family cytokines that encompass IL-1, IL-18 and IL-36 [9][25] and is constitutively expressed in the nucleus of non-immune cells, more particularly in endothelial and epithelial cells, fibroblasts and myofibroblasts [10][11][26,27]. Upon physiological conditions, IL-33 is localized in the nucleus, bound to chromatin (via the tails of histones H2A and H2B) [12][28], and acts as the keeper of epithelial barrier integrity through its transcriptional regulation abilities [13][14][15][29,30,31]. In pathological settings, if a breach in the epithelial barrier occurs, ensuing mechanical stress-induced cell death or necrosis, IL-33 is passively released in the extracellular compartment where it acts as an alarmin or damage-associated molecular pattern (DAMP) [16][32]. Extracellular full-length IL-33 is processed by proteases derived from neutrophils [17][33] and mast cells [18][34], generating truncated forms displaying biological activity up to 30-fold higher than the full-length IL-33 [17][18][33,34]. Extracellular IL-33 exerts its functions through the receptor ST2 and its coreceptor IL-1 receptor accessory protein (IL-1RacP, also known as IL1-R3). Due to alternative splicing, three isoforms of ST2 have been described: the transmembrane receptor type (ST2L), the soluble form (sST2) and the variant ST2 (ST2V) [19][20][21][35,36,37]. The binding of IL-33 to the transmembrane receptor ST2 enables its dimerization with IL-1RacP, further activating intracellular signaling through the myeloid differentiation primary response 88 (MyD88) adaptor, interleukin receptor-associated kinase (IRAK)1, IRAK4 and tumor necrosis factor receptor-associated factor (TRAF)6. This enables the activation of mitogen-activated protein (MAP) kinases and the nuclear factor κB (NFκB) transcription factor, leading to cell proliferation and the secretion of pro-inflammatory cytokines such as IL-4, IL-5 and IL-13 [22][23][38,39] (Figure 1).Figure 1.
IL-33/ST2 axis signaling.
The IL-33/ST2 axis is tightly regulated at several levels. sST2 acts as a decoy receptor of IL-33 and prevents its interaction with ST2, thereby counteracting its systemic effects [24][40]. The IL-33/ST2 axis is also antagonized by the single immunoglobulin domain IL-1R-related molecule (SIGGIR; also known as TIR8) that splits the heterodimer ST2/IL-1RacP, and by the activation of the ubiquitin–proteasome system, which digests ST2 [25][26][41,42]. Once released in the extracellular environment, inactivation of IL-33 occurs rapidly after approximatively 2 h through the oxidation of its cysteine residues and the formation of disulfide bridges [27][43].
The IL-33/ST2 axis mediates the activation of both myeloid and lymphoid cells and induces mainly a type 2 immune response, through the secretion of Th2-type cytokines (IL-5 and IL-13) and Th2 cell polarization [28][29][44,45]. ST2 is found in a wide variety of immune cells, including mast cells [30][46], basophils [31][47], eosinophils [32][48], M2 macrophages [33][49], neutrophils [31][47], NK cells [7][19], innate NK (iNK) cells [7][19], ILC2 [34][50], Treg [35][51] and Th2 cells [28][44]. However, under specific circumstances, IL-33 can also promote type 1 and type 17 immune responses [36][52]. More specifically, the production of type I IFN has been demonstrated following IL-33 exposure, leading to ST2 activation in type 1 helper (Th1) cells, NK cells and CD8+ T cells [7][8][37][19,20,53]. In asthma mouse models, the IL-33/ST2 activation in mast cells triggered a Th17 immune response [37][53]. In vitro studies on mouse macrophages showed that IL-33 exposure increased the expression of TLR-4, myeloid differentiation protein (MD)-2 and MyD88 [38][54]. In mouse bone marrow-derived DCs, the activation of ST2 upon IL-33 exposure increased the expression of DC maturation markers (CD80, CD40), pro-inflammatory cytokines (IL-4, IL-5, IL-13, tumor necrosis factor (TNF)-α and IL-1β) and chemokines such as C-C motif chemokine ligand 17 (CCL17) [39][55]. Therefore, IL-33 is a potent initiator of the innate immune response and can further activate adaptive immunity.
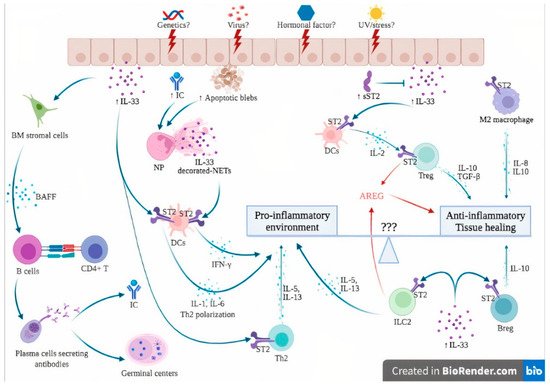
3. Expression of IL-33 and ST2 in Systemic Lupus Erythematosus
The IL-33/ST2 axis has been recently incriminated in the pathogenesis of SLE, but its precise contribution still remains elusive, partly due to the lack of clinical studies. The human IL-33 gene is located on chromosome 9p24.1 in humans [40][78]. The association between IL-33 gene polymorphisms and SLE has been studied exclusively in the Chinese population. Two polymorphisms, the rs1929992-G and rs1891385-C alleles, have been linked to the risk of SLE [41][42][43][79,80,81]. However, the increase was only moderate, with an odds ratio of 1.4 to 1.6 for the rs1891385C allele [41][42][79,80], and 1.3 to 1.6 for the rs1929992-G allele [42][43][80,81]. In addition, IL-33 serum levels of SLE patients only correlated with the rs1891385C allele [41][79]. Conflicting data exist regarding the serum levels of IL-33 in SLE patients. It was reported in several studies that IL-33 levels were significantly increased in the serum of patients with SLE compared with healthy controls [41][44][45][46][47][21,79,82,83,84]. IL-33 levels correlated with the disease activity score (Systemic Lupus Erythematosus Disease Activity Index, SLEDAI) [47][84] and acute inflammatory parameters such as the erythrocyte sedimentation rate (ESR) and C reactive protein (CRP), suggesting a potential interest for its use as a surrogate marker in the acute phase of SLE [45][82]. In contrast, sST2 serum levels have been more consistently reported to be significantly elevated across studies and correlated with the disease activity score (SLEDAI) [44][48][49][21,61,88] and with anti-double-stranded deoxyribonucleic acid (anti-dsDNA) antibodies [50][51][60,86].4. The Pathophysiological Role of IL-33 in Systemic Lupus Erythematosus
Preclinical studies report conflicting data regarding the role of the IL-33/ST2 axis in the pathophysiology of systemic lupus erythematosus. In the lupus-prone model of MRL/Lpr mice, anti-IL-33 treatment from weeks 14 to 20 significantly reduced mortality and lessened serum anti-dsDNA levels and circulating ICs. Renal biopsies showed alleviated renal damage as suggested by the reduced score of glomerulonephritis (GN), reduced renal IC deposition and reduced proteinuria. Finally, anti-IL-33 antibody treatment promoted the expansion of Treg and myeloid-derived suppressor cells (MDSCs) and decreased pro-inflammatory cytokines such as IL-17, IL-1β and IL-6. These data suggest that IL-33 antagonization has a protective effect on SLE [47][84]. In addition, results from WT mice chronically exposed to IL-33 showed a dramatic increase in B-cell activating factor (BAFF) levels, leading to the production of B and T follicular helper cells, the apparition of germinal centers and the apparition of IgG anti-DNA antibodies. These data suggest the potential involvement of IL-33 as a link between innate and adaptive immunity, and as a potent breaker of immune tolerance through IL-33-mediated BAFF production [49][88]. Conversely, the effect of early IL-33 administration in lupus-prone NZB/W F1 mice from weeks 6 to 12 significantly reduced proteinuria and mortality. Histological analysis revealed a significant reduction in glomerular and tubular damage scores and less deposition of ICs. IL-33 treatment also promoted IgM anti-dsDNA antibodies, IL-10-positive Breg cells and an M2 macrophage gene signature according to RNA sequencing data. These data suggest that IL-33 may exert a protective role during the development of SLE [52][90]. It has to be underscored that there is an antibody-independent production of B cells relating to autoimmunity, and that lupus mice with B cells unable to produce autoantibodies developed a lessened form of nephritis relative to those without B cells. The mechanisms of the protective role of IgM autoreactive anti-dsDNA antibodies in lupus nephritis are not fully deciphered but could be explained by the significant reduction in the production of pro-inflammatory cytokines such as TNF-α and IFNγ. It has been postulated that, in opposition to IgG anti-dsDNA antibodies harboring a cardinal role in fostering inflammation through the production of inflammatory cytokines by macrophages in the kidneys, IgM antibodies might lessen the inflammatory environment and inhibit the formation of immune complexes [53][54][91,92]. The hypothetical involvement of the IL-33/ST2 axis in the pathogenesis of SLE is further detailed in Figure 23. In genetically susceptible subjects exposed to a wide range of environmental factors such as viruses, UV light and stress [55][3], the products of cell damage arising from injured epithelial barriers lead to the passive release of IL-33. sST2 levels rise in an attempt to counteract the sudden increase in extracellular IL-33, as suggested by immunostaining from patients with lupus nephritis, where an increase in both IL-33 and sST2 was observed [44][21].Figure 23. Hypothesis of the involvement of the IL-33/ST2 axis in the pathogenesis of systemic lupus erythematosus. The dual role of the IL-33/ST2 axis can be seen as a balance between pro-inflammatory and anti-inflammatory effects. Unknown factors (3 questions marks in the figure) can influence this balance, skewing the immune response toward either pro- or anti-inflammatory states. In genetically susceptible subjects, environmental stimuli such as viruses, UV light and stress may trigger cell death and necrosis of the epithelial barrier, leading to passive release of IL-33, apoptotic blebs and exposure of autoantigens, ultimately leading to the formation of ICs. The products of cell damage, together with ICs, activate neutrophils to produce NETs that complex with IL-33 to activate DCs via their ST2 receptor, leading to a potent type I IFN secretion that contributes to the IFN signature of SLE. In addition, IL-33 also directly activates ST2 expressed by DCs, leading to the Th2 polarization of CD4+ T cells. IL-33 induces BAFF secretion by bone marrow stromal cells and possibly other, but not yet identified, cells that induce B cell differentiation into plasma cells, further contributing to germinal center formation and IC formation. Under certain conditions, probably in the early phase of the disease, the anti-inflammatory effects of IL-33 are dominant. sST2 levels are elevated to counteract IL-33 actions. IL-33/ST2 also induces IL-2 secretion by mast cells and dendritic cells, leading to Treg expansion. In addition, ST2 has been demonstrated on Treg, Breg and M2 macrophages, leading to anti-inflammatory cytokine production (IL-10, TGF-beta). Finally, ILC2 and Treg are also a source of AREG, which promotes tissue healing. Abbreviations: AREG: amphiregulin; BAFF: B-cell activating factor; Breg: regulatory B cells; BM: bone marrow; DCs: dendritic cells; IC: immune complexes; IFN-γ: interferon gamma; IL-: interleukin; ILC2: innate Lymphoid Cells type 2; NETs: neutrophil extracellular traps; NP: neutrophils; sST2: soluble ST2; ST2: receptor suppression of tumorigenicity 2; TGF-β: transforming growth factor beta; Th2: type 2 helper cells; TNFα: tumor necrosis factor alpha; Treg: regulatory T cells; UV: ultraviolet; ⊥: inhibit; blue and red ↓: induce/activate.